EINFÜHRUNG
Interstitielle Lungenerkrankungen bilden eine äußerst heterogene Gruppe von Erkrankungen, denen das Vorhandensein einer entzündlichen Komponente des Lungenparenchyms gemeinsam ist, die sich schließlich zu einer Fibrose entwickeln kann und die aus funktioneller Sicht eine einschränkende Veränderung verursacht, die schließlich zu Atemversagen führen kann.
Auch wenn es sich um Krankheiten handelt, deren Diagnose und Behandlung im Fachgebiet der Pneumologie erfolgt, wird die Behandlung grundsätzlich ambulant durchgeführt, und da es dabei zu verschiedenen Komplikationen kommen kann und oft auch kommt, ist es wichtig, dass der Hausarzt die aktuellen Leitlinien für die Behandlung dieser Krankheiten kennt, da er häufig von Patienten mit dieser Art von Problemen konsultiert wird, sei es aufgrund von Komplikationen der Atemwege oder anderer Art.
Der Zweck dieser Übersicht ist es, die therapeutischen Modalitäten, die diesen Patienten heute angeboten werden können, zu aktualisieren. Wir sind der Meinung, dass es nicht nur für den Hausarzt von größter Bedeutung ist, über ein aktuelles Wissen zu diesem Thema zu verfügen, sondern auch mit dem Pulmologen zusammenzuarbeiten, um eine gute Therapietreue des Patienten und eine frühzeitige Erkennung und Behandlung möglicher Komplikationen zu erreichen.
Unter den interstitiellen Erkrankungen ist die idiopathische Lungenfibrose (IPF) aufgrund ihrer Häufigkeit und ihres Schweregrades die wichtigste. Andererseits ist ihre Behandlung das Modell, auf dem die Behandlung fast aller dieser Krankheiten beruht, mit Ausnahme derjenigen, bei denen ein ätiologischer Erreger bekannt ist, da bei diesen die Beseitigung dieses Erregers die grundlegende Grundlage der Therapie ist. Wir werden uns daher auf eine Überprüfung der aktuellen Leitlinien für die Behandlung der IPF konzentrieren. Wir werden zunächst einen kurzen Überblick über die klinischen Merkmale geben und dann die aktuelle Behandlung der IPF im Detail erläutern.
KLINISCHE BESCHREIBUNG
Die IPF stellt wahrscheinlich den aggressivsten Prototyp der diffusen interstitiellen Pneumopathien dar. Die Prävalenz wird von einigen Autoren auf 5 Fälle pro 100.000 Einwohner geschätzt1, obwohl sie in Wirklichkeit unbekannt ist. Ihre Pathogenese hängt mit Veränderungen in den komplexen Bahnen des Immunsystems zusammen2,3.
Die häufigste klinische Erscheinung ist eine schleichend einsetzende Dyspnoe, die von einem trockenen Husten begleitet wird, der aufgrund seiner paroxysmalen und atemlosen Natur für den Patienten das lästigste Symptom sein kann. Im Allgemeinen ist die Dyspnoe zunächst unauffällig, und für eine frühzeitige Diagnose ist ein hoher Verdachtsindex erforderlich. Gelegentlich kommt es zu einem raschen Verlauf, der innerhalb weniger Monate zu einer irreversiblen Ateminsuffizienz führt, wie es für die ersten Beschreibungen von Hamman und Rich charakteristisch war. Häufiger ist jedoch, dass die Erkrankung zum Zeitpunkt des ersten Besuchs des Patienten bereits seit Monaten fortgeschritten ist.
Bei der körperlichen Untersuchung sind feine, trockene Knistergeräusche (Kehlkopfgeräusche) sehr charakteristisch und können in mehr als 80 % der Fälle auskultiert werden. In fortgeschrittenen Fällen kann es zu Atemnot bei leichter Anstrengung oder sogar zu Lippenzyanose kommen. Akropaquien können in etwa 30-50 % der Fälle auftreten. Der Verdacht auf IPF erhärtet sich, wenn auf dem Röntgenbild und der Computertomographie des Brustkorbs Anzeichen wie netzartige Muster, Bereiche mit Bodenglasinfiltration und ein vermindertes Lungenvolumen zu erkennen sind. Schließlich wird die Funktionsprüfung das Vorliegen eines restriktiven Musters bestätigen, und die arterielle Blutgasanalyse wird das Vorliegen einer Hypoxämie bestätigen. In Zweifelsfällen ist der Nachweis einer Hypoxämie bei Anstrengung von großem Wert.
Die endgültige Bestätigung der Diagnose kann nur durch eine Lungenbiopsie erfolgen. In der Realität überwiegen jedoch selbst in vielen der veröffentlichten Serien Fälle ohne histologische Diagnose. Kürzlich hat die American Thoracic Society (ATS) vorgeschlagen, dass das Vorhandensein all dieser Kriterien bedeutet, dass bei fehlender Exposition gegenüber Medikamenten, Umwelteinflüssen oder Bindegewebserkrankungen die Diagnose IPF höchstwahrscheinlich auch ohne Lungenbiopsie gestellt werden kann4, wenn eine Lungenbiopsie ein inakzeptables Risiko darstellen würde.
Usual interstitial pneumonia (UIP) ist das histopathologische Muster, das diese Patienten identifiziert4. Diejenigen mit den Mustern einer desquamativen interstitiellen Pneumonie, einer interstitiellen Erkrankung in Verbindung mit einer respiratorischen Bronchiolitis, einer unspezifischen interstitiellen Pneumonie, einer lymphoiden interstitiellen Pneumonie, einer akuten interstitiellen Pneumonie und einer Bronchiolitis obliterans mit organisierender Pneumonie werden als unterschiedliche Entitäten betrachtet.
Sobald die Diagnose und der klinische Schweregrad feststehen, benötigen die meisten Patienten eine Behandlung, da es sich um eine tödliche Erkrankung handelt, die nicht spontan abklingt. Das Ansprechen auf die Behandlung ist gering. Die Fünfjahresüberlebensrate beträgt 50 %5,2,6-8. Die Behandlung dauert etwa 3-6 Monate, bevor die Wirksamkeit festgestellt wird. Berichte über die Behandlung dieser Krankheiten sind rar: unkontrollierte Therapieversuche mit meist kleinen Patientenzahlen9.
Trotz des unvermeidlichen schleichenden und unumkehrbaren Fortschreitens der Krankheit ist therapeutischer Nihilismus nicht gerechtfertigt. Es gibt Beispiele für eine unbehandelte Fibrose mit langsamem Fortschreiten, die zu einer irreversiblen Fibrose führt, die nicht auf eine Behandlung anspricht oder eine schnell fortschreitende Phase entwickelt. Alle erfordern wahrscheinlich eine Behandlung zum Zeitpunkt der Vorstellung. Patienten, bei denen eine Behandlung kontraindiziert ist oder bei denen keine Anzeichen für eine funktionelle Beeinträchtigung vorliegen, können jedoch in Abständen von 3 bis 6 Monaten ein Folgeprogramm absolvieren. Auch die Behandlung älterer Patienten mit überwiegendem Fibrosebefall und schwerer Lungenfunktionseinschränkung ist schwierig. Diese Patienten erhalten eine kurze Behandlung von 3 bis 6 Monaten, um die Reversibilität festzustellen. Nur wenige von ihnen werden positiv reagieren.
Es ist oft schwierig festzustellen, ob die Behandlung die gewünschte Wirkung zeigt. Häufig tritt eine subjektive Verbesserung ein (bei bis zu 70 % der Patienten). Eine objektive Verbesserung tritt bei 20-30 % ein. Die am häufigsten verwendeten Parameter zur Definition einer funktionellen (objektiven) Verbesserung sind eine Zunahme der Gesamtlungenkapazität (TLC) oder der Vitalkapazität (VC), eine Zunahme der Diffusionskapazität und eine Verringerung oder Normalisierung des SaO2-Abfalls während der Belastung4,10-12.
Behandlung
Kortikosteroide
Kortikosteroide sind die Hauptstütze der Behandlung von IPF, obwohl ihr Erfolg eher gering ist: ein günstiges Ansprechen erfolgt bei einem Drittel der Patienten. Der Grund für ihre Verwendung ist die Unterdrückung der chronischen Alveolitis. Man nimmt an, dass sie die Migration von Neutrophilen und Lymphozyten in der Lunge unterdrücken, indem sie die Konzentration von Immunkomplexen verringern, die Funktion der Alveolarmakrophagen und die Adhäsion von Neutrophilen an Endotheloberflächen verändern.
Es ist nicht klar, warum manche Patienten darauf ansprechen und andere nicht. Dies könnte mit dem Vorhandensein von Oberflächenrezeptoren auf Zellen des Lungenparenchyms zusammenhängen. Ein kurzer Verlauf der Symptome, leichte funktionelle und radiologische Beeinträchtigungen, weibliches Geschlecht und jüngeres Alter der Patienten scheinen mit einem besseren Ergebnis verbunden zu sein.
Obwohl es sich um die am weitesten verbreitete Behandlung handelt, wurde ihre langfristige Wirksamkeit in keiner prospektiven, einfach verblindeten, placebokontrollierten Studie bestätigt.
Izumi et al. analysierten das Überleben von 222 Patienten und stellten fest, dass es nach 10 Jahren keinen Unterschied im Überleben zwischen denjenigen gab, die Steroide erhalten hatten, und den unbehandelten Patienten13. Wenn ein Patient mit IPF jedoch eine eingeschränkte Lungenfunktion oder Symptome aufweist, die sein normales Leben beeinträchtigen, ist es sinnvoll, einen Versuch einer Kortikosteroidtherapie einzuleiten. Eine spontane Besserung der Krankheit ist sehr selten, und wenn man sich entschließt, den Beginn der Behandlung hinauszuzögern, sollte dies auf Kosten einer genauen Überwachung des Patienten geschehen, damit die Behandlung eingeleitet werden kann, sobald sich eine Verschlechterung abzeichnet.
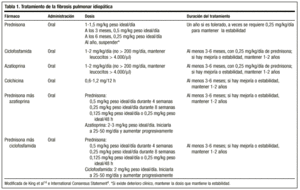
Die klassische Behandlung beginnt mit Prednison 1,5-2 mg/kg Idealgewicht/Tag als Einzeldosis am Morgen, wobei eine Dosis von 100 mg nicht überschritten werden sollte. Nach 2-3 Monaten wird der Patient durch Röntgenaufnahmen und Funktionsuntersuchungen erneut untersucht. Spricht der Patient darauf an, wird Prednison (1-2 mg/Woche) auf eine Erhaltungsdosis von 0,5 mg/kg Idealgewicht/Tag reduziert, die dann weiter gesenkt werden kann. Wenn keine Reaktion erfolgt, wird ein Immunsuppressivum verabreicht. Im Allgemeinen ist es unwahrscheinlich, dass die Behandlung abgebrochen werden kann, und Dosen von 0,25 mg/kg Idealgewicht/Tag werden normalerweise beibehalten.
In jüngster Zeit haben sich die Amerikanische und die Europäische Thoraxgesellschaft darauf geeinigt, die Behandlung mit einer Kombination aus Prednison oder einem anderen Kortikosteroid in gleicher Dosierung und einem Immunsuppressivum (Azathioprin oder Cyclophosphamid)4 zu beginnen, um die Nebenwirkungen dieses Medikaments zu vermeiden und bessere Überlebensraten zu erzielen: Prednison 0,5 mg/kg Idealgewicht/Tag für 4 Wochen; 0,25 mg/kg Idealgewicht/Tag für 8 Wochen und dann Verjüngung auf 0,125 mg/kg Idealgewicht/Tag oder 0,25 mg/kg Idealgewicht/48 h.
Es gibt keine Leitlinien für die Dauer der Behandlung; die Mindestdauer beträgt ein Jahr. Es ist möglich, dass die Krankheit nach Monaten oder Jahren reaktiviert wird; in diesem Fall sollte eine Behandlung mit denselben Mitteln eingeleitet werden. In jedem Fall sollte das Ansprechen auf die Behandlung immer anhand einer objektiven Verbesserung bewertet werden, und die Möglichkeit, den Patienten chronisch auf einer Dosis zu halten, die Stabilität garantiert, sollte den Fällen vorbehalten bleiben, in denen eine Verbesserung mit Kortikosteroiden, eine Verschlechterung beim Absetzen und eine neue Verbesserung bei erneuter Erhöhung der Dosis auftritt. Die für jeden Patienten erforderliche Dosis kann nur von Fall zu Fall in Abhängigkeit von der Reaktion auf Dosisänderungen bestimmt werden. Viele Autoren halten eine lebenslange Behandlung für notwendig.14
Wenn die Krankheit anfänglich schwer verläuft, werden hochdosierte Kortikosteroide vorgeschlagen. Keogh et al. haben bei Patienten mit schwerer Erkrankung intermittierend hochdosierte parenterale Kortikosteroide eingesetzt: intravenöses Methylprednisolon, 2 g einmal wöchentlich, plus orales Prednison, 0,25 mg/kg/Tag. Der Grund dafür ist, dass sie eine stärkere Wirkung auf die neutrophile Alveolitis (die mit einer schlechten Prognose verbunden ist) haben als niedrig dosierte Steroide15. Diese Arbeit weist jedoch einige Mängel auf, wie z. B. die kurze Nachbeobachtungszeit von 6 Monaten, die geringe Anzahl von Patienten und die unter der Norm liegenden Prednisondosen9. Die anfängliche Behandlung mit hochdosierten intravenösen Kortikosteroiden (Methylprednisolon 250 mg/6 h) wurde eingesetzt, um die Krankheit so früh wie möglich unter Kontrolle zu bringen, wenn sie schnell fortschreitet. Es gibt keine kontrollierten Studien zur Wirksamkeit dieser Methoden; höhere Dosen scheinen jedoch eine größere Wirksamkeit zu haben, obwohl auch die Nebenwirkungen größer sind.14.
ZITOTOXISCHE WIRKSTOFFE
Sie wurden klassischerweise als Alternative zu Kortikosteroiden eingesetzt, wenn diese unwirksam waren und auch, wenn ihre Nebenwirkungen ihre weitere Verabreichung ausschließen. Im Einklang mit dem äußerst praktischen Geist dieses Artikels werden wir die Wirkstoffe besprechen, die sich in der klinischen Praxis als nützlich erwiesen haben, obwohl uns, wie im Falle der Kortikosteroide, lange, prospektive und randomisierte Serien fehlen, die es uns erlauben, ihre genaue Rolle bei der Behandlung der IPF zu kennen.
Azathioprin
Sein Wirkmechanismus beruht auf der Substitution von Purinen in der Desoxyribonukleinsäure-Synthese, wodurch die Adenin-Desaminase gehemmt und die Lymphozyten inaktiviert werden. Weitere Wirkungen sind die Unterdrückung der Aktivität natürlicher Killerzellen, der Antikörperproduktion und der antikörperabhängigen zellulären Zytotoxizität.
Die empfohlene Dosis beträgt 2 mg/kg/Tag, nicht mehr als 200 mg/Tag, über einen Zeitraum von 3-6 Monaten. Die Verabreichung beginnt mit einer Dosis von 50 mg/Tag und wird bei guter Verträglichkeit in Abständen von 1 bis 2 Wochen erhöht, bis die geplante Dosis erreicht ist.
Die wichtigsten Nebenwirkungen sind hämatologischer Natur: Leukopenie, Anämie, Thrombozytopenie, Erythrozytenaplasie und Panzytopenie. Die Leukozytenzahl muss überwacht werden. Sinkt sie unter 4.000/μl, muss Azathioprin abgesetzt werden, bis sie wieder ansteigt. Darüber hinaus können Magen-Darm-Beschwerden wie Übelkeit und Erbrechen, Magengeschwüre, Durchfall und eine leichte Erhöhung der Leberenzyme auftreten. Insgesamt erfordern die Nebenwirkungen in bis zu 20-30 % der Fälle ein Absetzen von Azathioprin.
Raghu et al. verglichen die Wirksamkeit von hochdosierten Glukokortikoiden mit der von Azathioprin in Kombination mit niedrig dosierten Glukokortikoiden. Die Anwendung dieses Medikaments ging mit einer bescheidenen Verbesserung der Atemfunktionsparameter und einer Verlängerung der Überlebenszeit einher17.
Cyclophosphamid
ist ein Alkylierungsmittel. Seine Wirkungsweise ist die Verarmung der Lymphozyten und unterdrückt somit die Lymphozytenfunktion. Die empfohlene Dosis beträgt 2 mg/kg/Tag einzeln, oral, in der Regel in Kombination mit 0,25 mg/kg/Tag oralem Prednison, und darf 200 mg/Tag für mindestens 3 Monate und bis zu 9-12 Monaten nicht überschreiten.
Die wichtigste Nebenwirkung ist Leukopenie. Die Gesamtleukozytenzahl muss größer als 3.000/μl oder die Neutrophilenzahl größer als 1.500/μl sein. Weitere Komplikationen sind Thrombozytopenie, Hämaturie infolge hämorrhagischer Zystitis, Anorexie, Übelkeit und Erbrechen, Knochenmarksuppression, Azoospermie und Amenorrhoe, Infektionen und hämatologische Malignität.
Kolb et al. untersuchten 18 Patienten mit IPF, um die Wirksamkeit und Sicherheit der gepulsten Cyclophosphamidtherapie zu ermitteln. Cyclophosphamid wurde intermittierend (1-1,3 g/Monat) zusammen mit oralem Prednisolon ein Jahr lang verabreicht. Nach diesem Zeitraum wurde Cyclophosphamid abgesetzt und die erreichte Kortikosteroiddosis fortgesetzt8 . Sie wurden mindestens 14 Monate lang beobachtet, wobei Lebensqualität, TLC, FEV1, CO-Transferkapazität (DLCO), SaO2 sowie Symptome überwacht wurden. Bei einem Patienten musste die Behandlung wegen rezidivierender Lungenentzündungen abgebrochen werden; 11 Patienten galten als Responder (5 verbesserten und 6 stabilisierten sich) und 6 verschlechterten sich. Die Langzeittoxizität von Cyclophosphamid hängt von der kumulativen Dosis ab. Intermittierende Dosen sind ebenso wirksam und besser verträglich als tägliche Dosen. Responder hatten einen höheren Anteil an Lymphozyten in der BAL. Ein weiteres bemerkenswertes Ergebnis ist, dass die anfängliche Lebensqualität der Responder aufgrund der kürzeren Zeit bis zum Fortschreiten der Krankheit signifikant besser war, was darauf hindeutet, dass diese Behandlung frühzeitig begonnen werden sollte.
Baughman und Lower behandelten 33 IPF-Patienten 18 Monate lang mit 1.000-1.800 mg Cyclophosphamid/2 Wochen. Diejenigen, die 18 Monate überlebten, wiesen eine signifikante Verbesserung der VCF auf, die auch im folgenden Jahr anhielt. Die Prednisolonmenge konnte von 32 auf 4 mg/Tag reduziert werden16.
Penicillamin
Antifibrosivum. Es wird vermutet, dass es durch die Beeinträchtigung der Bildung von intra- und intermolekularen kovalenten Bindungen der Kollagenfasern die Entwicklung der Lungenfibrose hemmt. Es wurden einige Fälle von Bindegewebserkrankungen mit begleitender Lungenfibrose beschrieben, bei denen es eine gewisse Wirksamkeit gezeigt hat, aber es gibt keine kontrollierten Studien mit großen Patientenserien, so dass es derzeit nicht als gültige Behandlungsalternative angesehen werden kann. Daher sind weitere Studien erforderlich, bevor eine Empfehlung ausgesprochen werden kann.
Colchizin
Verringert die Kollagensekretion und erhöht die kollagenolytische Aktivität. Es hat auch eine entzündungshemmende Wirkung. Es sind noch Studien erforderlich, um seine Rolle bei der Behandlung dieser Krankheit genauer zu bestimmen, aber es gibt Studien, in denen ihm eine ähnliche Wirksamkeit wie den Kortikosteroiden zugeschrieben wurde, und seine Nebenwirkungen sind nicht schwerwiegend. Es wird in einer Dosis von 0,6 mg ein- oder zweimal täglich bei Patienten empfohlen, die refraktär auf Kortikosteroide, allein oder in Kombination mit Immunsuppressiva, reagieren. Obwohl es Autoren gibt, die sie in die erste Behandlungslinie einordnen, fehlen uns derzeit Daten, um sie als solche zu etablieren (Tabelle 1).
PULMONAREN-TRANSPLANTATION
Fälle, in denen die Krankheit ohne Ansprechen auf die Behandlung fortschreitet und schließlich zum Atemversagen führt, sind bei dieser Krankheit leider am häufigsten. Für diese Patienten gibt es derzeit keine Aussicht auf eine medizinische Behandlung außer der Palliation. Vor diesem düsteren Hintergrund kam in den 1980er Jahren die Möglichkeit der Lungentransplantation auf und hat sich so weit entwickelt, dass sie heute eine therapeutische Option darstellt, die als Instrument für den klinischen Einsatz in Betracht gezogen werden sollte und nicht mehr auf den experimentellen Bereich beschränkt ist. Am besten geeignet ist es für Menschen unter 65 Jahren, bei denen keine fortschreitende und irreversible Erkrankung anderer Organe sowie keine aktive Infektion oder Infektion mit multiresistenten Keimen vorliegt. Eine angemessene persönliche Veranlagung und ein guter Ernährungszustand sind ebenfalls erforderlich. Patienten mit einer CPT von weniger als 60 % des erwarteten Wertes oder einer Lungen-Diffusionskapazität von weniger als 40 % haben eine Überlebenszeit von weniger als 2 Jahren. Bei diesem Grad der Funktionsstörung benötigen sie eine Sauerstofftherapie und entsättigen leicht bei minimaler Bewegung. In diesen Fällen sollte eine Lungentransplantation in Erwägung gezogen werden, sobald eine Sauerstofftherapie notwendig wird und ein unaufhaltsames Fortschreiten der Krankheit nachgewiesen ist. Die bevorzugte Transplantationsart ist die Ein-Lungen-Transplantation, während die Zwei-Lungen-Transplantation für Fälle reserviert ist, in denen Zweifel über das Verhalten der Restlunge bestehen. Wenn der Patient weniger als 50 Jahre alt ist und eine Rechtsherzinsuffizienz hat, kann eine Herztransplantation in Betracht gezogen werden. In diesem Fall ist die Krankheit jedoch bereits sehr weit fortgeschritten und es ist unwahrscheinlich, dass sie die Wartezeit übersteht18,19. Die Überlebensrate nach der Transplantation liegt im ersten Jahr bei 80 % und nach 5 Jahren bei 50-60 %, was die Prognose für einen Patienten mit IPF, der eine Ateminsuffizienz erreicht hat, erheblich verbessert.
Bewertung des Ansprechens auf die Behandlung
Wenn keine Komplikationen auftreten, kann es bis zu 6 Monate dauern, bis ein Ansprechen auf die Behandlung erfolgt. Hat sich die Situation des Patienten nach dieser Zeit gebessert oder ist er stabil, sollten die gleichen Dosen beibehalten werden. Wenn sie sich verschlimmert haben, sollten sie auf eine alternative Therapie (ein anderes Immunsuppressivum oder sogar eine Transplantation) umgestellt werden. Nach 12 Monaten sollte eine erneute Bewertung auf die gleiche Weise erfolgen, und nach 18 Monaten sollte sie je nach Reaktion individuell angepasst werden. Se recomienda continuar indefinidamente sólo en aquellos pacientes con mejoría continua o estabilización (tabla 2).

Una respuesta favorable se define por dos o más de los siguientes criterios en dos visitas consecutivas:
1. Mejoría de los síntomas, sobre todo en el grado de esfuerzo requerido para que el paciente deba parar cuando hace un esfuerzo o un descenso en la frecuencia o gravedad de la tos.
2. Reducción de las alteraciones radiológicas.
3. Mejoría funcional definida por dos o más de los siguientes parámetros:
Aumento >= 10% en la TLC o CV (o >= 200 ml).
Aumento >= 15% en DLCO o >= 3 ml/min/mmHg.
Mejoría o normalización de la SaO2 o pO2 (aumento >= 4% o >= 4 mmHg) durante un prueba de esfuerzo cardiorrespiratoria.
Una respuesta estable (también considerada favorable) se define por dos o más de los siguientes criterios en dos visitas consecutivas:
1. Cambio de
2. Cambio de
3. Sin cambios en la SaO2 o pO2 (aumento
Un fracaso del tratamiento se define por dos o más de los siguientes criterios en dos visitas consecutivas:
1. Aumento de los síntomas, sobre todo disnea o tos.
2. Aumento de las alteraciones radiológicas, especialmente imágenes en panal o signos de hipertensión pulmonar.
3. Deterioro funcional definido por dos o más de los siguientes parámetros:
Descenso >= 10% en la TLC o CV (o >= 200 ml).
Descenso >= 15% en DLCO o >= 3 ml/min/mmHg.
Verschlechterung des SaO2 (= 4%) oder Erhöhung des alveolo-arteriellen Gradienten (AapO2) (= 4 mmHg) in Ruhe oder während eines kardiorespiratorischen Belastungstests.
ZUSAMMENFASSUNG
Diopathische Lungenfibrose ist eine tödliche Krankheit, die nicht spontan abklingt und schlecht auf die Behandlung anspricht. Zu letzterem gibt es nur wenige Studien mit einer kleinen Anzahl von Patienten. Kortikosteroide, die mindestens ein Jahr lang in unterschiedlichen Dosierungen verabreicht werden, sind die gängigste Behandlungsmethode, allein oder in Kombination mit anderen Immunsuppressiva. Kürzlich wurde die Kombinationstherapie von der American Thoracic und der European Respiratory Society4 befürwortet. Da viele Patienten erst in späten Stadien diagnostiziert werden, wird an Antifibrosemitteln geforscht, die verhindern sollen, dass sich der fibrotische Prozess etabliert und damit irreversibel wird. Angesichts der Überlebensraten und der Entwicklung sollte eine Lungentransplantation in Betracht gezogen werden, sobald die Behandlung versagt und die Patienten eine Sauerstofftherapie benötigen. All dies deutet auf die Notwendigkeit hin, multizentrische Studien durchzuführen, um eine ausreichende Anzahl von Patienten zu rekrutieren, die Wirksamkeit der verabreichten Behandlungen zu bewerten und den natürlichen Verlauf der Krankheit sowie die tatsächliche Inzidenz und Prävalenz zu ermitteln4,20. Andererseits wird empfohlen, die Krankheit in einem frühen Stadium zu erkennen und eine Behandlung einzuleiten, um höhere Überlebensraten zu erzielen.