Viele der Methoden zur Visualisierung und Interpretation von Genexpressionsdaten können sowohl für Microarray- als auch für RNA-seq-Experimente verwendet werden. Einige der gebräuchlichsten Methoden werden im Folgenden erörtert.
Heatmaps und Clustering
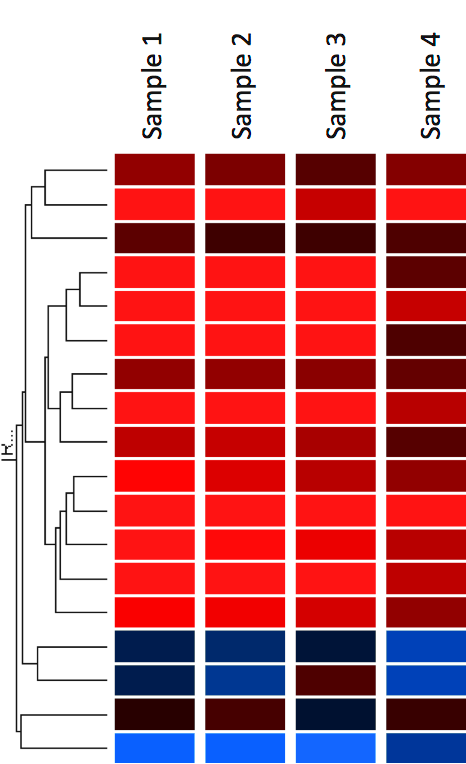
Eine gängige Methode zur Visualisierung von Genexpressionsdaten ist die Darstellung als Heatmap (Abbildung 12). Die Heatmap kann auch mit Clustering-Methoden kombiniert werden, die Gene und/oder Proben auf der Grundlage der Ähnlichkeit ihrer Genexpressionsmuster gruppieren. Dies kann nützlich sein, um Gene zu identifizieren, die gemeinsam reguliert werden, oder biologische Signaturen, die mit einem bestimmten Zustand (z. B. einer Krankheit oder einer Umweltbedingung) verbunden sind (4).
In Heatmaps werden die Daten in einem Raster dargestellt, wobei jede Zeile ein Gen und jede Spalte eine Probe darstellt. Die Farbe und Intensität der Kästchen wird verwendet, um Veränderungen (nicht absolute Werte) der Genexpression darzustellen. Im folgenden Beispiel steht Rot für hochregulierte Gene und Blau für herunterregulierte Gene. Schwarz steht für eine unveränderte Expression.
Analyse der Anreicherung von Gensätzen und Analyse von Signalwegen
Ein gängiger Ansatz zur Interpretation von Genexpressionsdaten ist die Analyse der Anreicherung von Gensätzen auf der Grundlage der funktionellen Annotation der unterschiedlich exprimierten Gene (Abbildung 13). Dies ist nützlich, um herauszufinden, ob die unterschiedlich exprimierten Gene mit einem bestimmten biologischen Prozess oder einer molekularen Funktion in Verbindung stehen.
Die Gene Ontology, die eine standardisierte Annotation von Genprodukten enthält, wird üblicherweise für diesen Zweck verwendet. Dabei wird die Häufigkeit einzelner Annotationen in der Genliste (z.B. differentiell exprimierte Gene) mit einer Referenzliste (meist alle Gene auf dem Microarray oder im Genom) verglichen. Die Anreicherung von biologischen Pfaden, die von KEGG, Ingenuity, Reactome oder WikiPathways geliefert werden, kann auf ähnliche Weise durchgeführt werden (12,13).
Populäre Tools für die Anreicherung von Gensätzen und die Analyse von Pfaden sind:
- DAVID (kostenloses Online-Tool)
- GSEA (kostenlos)
- Ingenuity (Lizenz erforderlich)
- Reactome (kostenlos)

Netzwerkanalyse
Die Netzwerkanalyse ergänzt die Pfadanalyse und kann dazu verwendet werden, um zu zeigen, wie Schlüsselkomponenten verschiedener Pfade interagieren. Dies kann nützlich sein, um regulatorische Ereignisse zu identifizieren, die mehrere biologische Prozesse und Pfade beeinflussen (12,13).