- Einführung
- Materialien und Methoden
- Probenvorbereitung
- MinION-Bibliotheksvorbereitung und Sequenzierung
- Genomics Analysis
- Ergebnisse
- MinION-Sequenzierungsdaten und Zusammenbau von Virusgenomen
- Ausmaß und Quelle der probenübergreifenden Kontamination
- Diskussion
- Beiträge der Autoren
- Finanzierung
- Erklärung zu Interessenkonflikten
- Dankesworte
- Zusatzmaterial
- Fußnoten
Einführung
Die metagenomische Sequenzierung hat das Potenzial, eine unvoreingenommene Identifizierung von Krankheitserregern aus einer klinischen Probe zu ermöglichen. Sie verspricht, als einziger und universeller Test für die Diagnostik von Infektionskrankheiten direkt aus Proben zu dienen, ohne dass a priori Wissen erforderlich ist (Bibby, 2013; Miller et al., 2013; Schlaberg et al., 2017). Neben der Identifizierung von Erregerspezies könnten breite und tiefe metagenomische Sequenzdaten Informationen liefern, die für die Festlegung von Behandlung und Prognose, die Erkennung von Ausbrüchen und die Verfolgung der Infektionsepidemiologie relevant sind (Greninger et al., 2010; Yang et al., 2011; Qin et al., 2012; Loman et al., 2013). Plattformen für die Sequenzierung der nächsten Generation (Next Generation Sequencing, NGS) können einen enormen Datendurchsatz zu bescheidenen Kosten erzeugen, doch ihre Anwendung in der klinischen Diagnostik und im öffentlichen Gesundheitswesen wurde durch Komplexität, Langsamkeit und Kapitalinvestitionen eingeschränkt.
Der MinION ist ein handtellergroßer Echtzeit-Einzelmolekül-Genomsequenzer, der von Oxford Nanopore Technologies (ONT) entwickelt wurde. Die kompakte Größe und der Echtzeit-Charakter des MinION könnten die Anwendung der metagenomischen Sequenzierung bei Point-of-Care-Tests für Infektionskrankheiten erleichtern, wie mehrere Proof-of-Concept-Studien zeigen, darunter die Identifizierung von Chikungunya (CHIKV), Ebola (EBOV) und Hepatitis-C-Virus (HCV) aus menschlichen klinischen Blutproben ohne Zielanreicherung (Greninger et al, 2015) und den Nachweis bakterieller Krankheitserreger aus Urinproben (Schmidt et al., 2016) und Atemwegsproben, ohne dass eine vorherige Kultur erforderlich ist (Pendleton et al., 2017).
Der Datendurchsatz von MinION hat sich seit seiner Veröffentlichung im Jahr 2015 stark erhöht, wobei jede verbrauchbare Fließzelle jetzt bis zu 10-20 Gb an DNA-Sequenzdaten erzeugt. Dies ermöglicht es den Nutzern, die Fließzelle effizienter zu nutzen (und die Kosten zu senken), indem mehrere Proben in einem einzigen Sequenzierungslauf gemultiplext werden. ONT hat PCR-freie Barcode-Sets entwickelt, die das Multiplexing von bis zu 12 Proben ermöglichen.
Der Nachweis von Influenza-A-Viren in mehreren Atemwegsproben könnte eine diagnostische Anwendung eines multiplexen MinION-Sequenzierungstests sein. Bei der direkten Sequenzierung von Proben mit einem potenziell breiten Spektrum an Virustitern ist es jedoch wichtig, sich des Potenzials einer Kreuzkontamination der Proben bewusst zu sein, sowohl während der Bibliotheksvorbereitung als auch in der bioinformatischen Barcode-Demultiplexierungsphase nach der Sequenzierung. Hier stellen wir einen einzigartigen MinION-Sequenzierungsdatensatz und die Ergebnisse einer Untersuchung des Ausmaßes und der Quelle der Kreuz-Barcode-Kontamination bei der Multiplex-Sequenzierung vor.
Materialien und Methoden
Wir haben eine mit dem Influenza-A-Virus infizierte Nasenspülprobe eines Frettchens als Beispiel verwendet und außerdem zwei Aliquots negativer Nasenspülproben von nicht infizierten Frettchen (bereits vorhandene unbenutzte Bestände aus einer nicht verwandten Studie) separat mit Dengue- und Chikungunya-Viren versetzt. Keines dieser Viren ist für die klinische Diagnostik in Atemwegsproben relevant, sondern dient hier als eindeutiger, klarer Marker für die Bewertung der Kreuzkontamination von Proben. Die Sequenzierbibliotheken für jede Probe wurden parallel zu einer negativen Nasenspülkontrolle hergestellt, mit einem Barcode versehen und einzeln sequenziert. Anschließend wurde ein Aliquot der Sequenzierbibliotheken gepoolt und eine Multiplex-MINION-Sequenzierung durchgeführt. Die Reads der vier Einzelläufe (bezeichnet als „CHIKV“, „DENV“, „FLU-A“ und „Negativ“) und des Multiplex-Laufs (bezeichnet als „Multiplexed“) wurden anschließend analysiert, um das Ausmaß und die Quelle der Kreuzprobenkontamination zu untersuchen.
Probenvorbereitung
Die Projektlizenz wurde vom örtlichen AWERB (Animal Welfare and Ethics Review Board) geprüft und anschließend vom Innenministerium erteilt. Die RNA wurde mit dem QIAamp-Kit für virale RNA (Qiagen) gemäß den Anweisungen des Herstellers aus Nasenspülungen von Frettchen, die Influenza-A(H1N1)-Viren (A/California/04/2009) enthielten, sowie aus einem Pool negativer Nasenspülproben extrahiert. Aliquots des negativen Probenextrakts wurden entweder mit Dengue (DENV) (Stamm TC861HA, GenBank: MF576311) oder CHIKV (Stamm S27, GenBank: MF580946.1) viraler RNA aus der Nationalen Sammlung pathogener Viren1 versetzt. Die Proben wurden mit TURBO DNase (Thermo Fisher Scientific, Waltham, MA, USA) DNase-behandelt und mit dem RNA Clean & ConcentratorTM-5-Kit (Zymo Research) gereinigt. cDNA wurde mit einer sequenzunabhängigen Single-Primer-Amplifikationsmethode (Greninger et al., 2015), die wie zuvor beschrieben modifiziert wurde (Atkinson et al., 2016), hergestellt und amplifiziert. Amplifizierte cDNA wurde mit dem Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA) quantifiziert, und 1 μg wurde als Input für jede MinION-Bibliotheksvorbereitung verwendet, mit Ausnahme der Negativkontrolle, bei der die gesamte Probe (32 ng) verwendet wurde.
MinION-Bibliotheksvorbereitung und Sequenzierung
Das Ligation Sequencing Kit 1D (SQK-LSK108) und das Native Barcoding Kit 1D (EXP-NBD103) wurden gemäß den ONT-Standardprotokollen verwendet, mit der Ausnahme, dass in jeder der vier Bibliotheksvorbereitungen nur ein Barcode enthalten war. Jede Bibliothek wurde auf einer individuellen Fließzelle ausgeführt, und eine fünfte gepoolte Bibliothek wurde durch Kombination der vier individuell barcodierten Bibliotheken hergestellt. Die Bibliotheken wurden auf R9.4-Fließzellen sequenziert. Das Studiendesign ist in Abbildung 1 dargestellt.
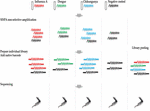
ABBILD 1. Überblick über das Studiendesign. RNA wurde aus vier Proben extrahiert, darunter eine mit dem Influenza-A-Virus infizierte Frettchen-Nasenspülprobe, zwei negative Frettchen-Nasenspülproben, die mit Dengue- und Chikungunya-Viren versetzt waren, und eine negative Frettchen-Nasenspülkontrolle. cDNA wurde hergestellt und mit einer sequenzunabhängigen Single-Primer-Amplifikationsmethode amplifiziert. Die Sequenzierungsbibliotheken für jede Probe wurden parallel hergestellt, mit einem Barcode versehen und auf einzelnen Durchflusszellen sequenziert. Die Multiplex-Sequenzierung wurde auch durch Pooling der vier Einzelbibliotheken durchgeführt. Die Reads aus den vier individuellen Läufen und dem Multiplex-Lauf wurden analysiert, um das Ausmaß und die Quelle der Cross-Barcode-Kontamination bei der Multiplex-Sequenzierung zu bewerten.
Genomics Analysis
Die Reads wurden mit Albacore v2.1.7 (ONT) mit Barcode-Demultiplexing als Basis aufgerufen. Die Reads aus jedem Sequenzierungslauf wurden mithilfe von Minimap2 (Li, 2018) auf die genomischen Sequenzen der einzelnen Viren abgebildet. Die Anzahl der auf die Referenz gemappten Reads wurde mit Pysam2 gezählt. Die De-novo-Assemblierung wurde mit Canu v1.7 (Koren et al., 2017) durchgeführt, und der resultierende Genom-Entwurf wurde mit Nanopolish (Mongan et al., 2015) mit den Signal-Level-Daten poliert.
Um ein stringentes Barcode-Demultiplexing der Multiplex-MinION-Sequenzierungsdaten zu ermöglichen, führten wir zwei Analyserunden mit Porechop (v0.2.23) durch. Das Vorhandensein einer Adaptersequenz in der Mitte eines Reads ist ein Zeichen für eine Chimäre. Mit Porechop untersuchten wir jeden Read, und diejenigen, deren mittlerer Bereich >75% Identität mit der Adaptersequenz aufweist, wurden als chimäre Reads identifiziert. In Porechop setzen wir die Option „-middle_threshold“ und wählen einen Schwellenwert von 75. In der zweiten Runde suchten wir mit Porechop sowohl am Anfang als auch am Ende eines Reads nach der Barcode-Sequenz; Reads wurden nur dann zugeordnet, wenn an beiden Enden der gleiche Barcode gefunden wurde. Wir setzten die Option „-require_two_barcodes“ in Porechop und setzten den Schwellenwert für die Barcodebewertung auf 70. Um eine mögliche Signatur der chimären Reads zu finden, untersuchten wir die in der FAST5-Datei des MinION-Sequenzers gespeicherten Read-Stromsignale. Die Stromsignale wurden mit ONT fast5 API4 extrahiert und mit Hilfe von ggplot2, das in R5 implementiert wurde, für einen Vergleich von chimären und nicht-chimären Reads aufgezeichnet.
Ergebnisse
MinION-Sequenzierungsdaten und Zusammenbau von Virusgenomen
Der Durchsatz jedes MinION-Sequenzierungslaufs variierte aufgrund der unterschiedlichen Laufzeiten. Eine maximale Anzahl von ∼2,4 Mio. Reads wurde durch den Multiplex-Sequenzierungslauf und den individuellen CHIKV-Lauf aufgrund längerer Laufzeiten erreicht (ergänzende Tabelle S1). Die Reads aus dem gespikten Virus machten 96 % der Daten in den einzelnen CHIKV- und DENV-Sequenzierungsläufen aus, und 78 % für die FLU-A-Probe (Tabelle 1). Der prozentuale Anteil der viralen Reads innerhalb jeder barcodierten Probe in den Multiplex-Sequenzierungsdaten ist ähnlich hoch wie in den individuell durchgeführten Probendaten (Tabelle 2). Jedes virale Genom hatte eine extrem hohe (>8.000) mittlere Abdeckungstiefe in den Einzel- und Multiplex-Sequenzierungsdaten, und die de novo-Assemblierung war in der Lage, nahezu vollständige Genome für alle drei Viren mit 99.

TABLE 1. Zusammenfassung der Mapping- und De-novo-Assembly-Ergebnisse für Daten aus der MinION-Sequenzierung einzelner Bibliotheken.

TABLE 2. Zusammenfassung der Mapping- und De-novo-Assembly-Ergebnisse für Daten aus der Multiplex-MINION-Sequenzierung.
Ausmaß und Quelle der probenübergreifenden Kontamination
Jede Probe wurde mit einem Barcode versehen und sowohl einzeln als auch im Multiplex sequenziert, was es uns ermöglichte, die Leistung der Barcode-Demultiplexierung von Albacore zu untersuchen. Bei den einzeln sequenzierten Probendaten würden wir erwarten, dass nur ein einziger nativer Barcode vorhanden ist. Bei den individuellen Sequenzierungsläufen von CHIKV (Barcode NB01), DENV (NB09) und FLU-A (NB10) stellten wir fest, dass 86, 109 bzw. 17 Reads Barcode-Bins zugewiesen wurden, von denen wir nicht erwartet hatten, dass sie in der Bibliothek vorhanden sind (dies entspricht 0,0036, 0,0129 bzw. 0,001 % der gesamten Reads). In den Multiplex-Sequenzierungsdaten wurden 41 Reads (0,0016 %) Barcodes zugewiesen, die nicht in den Experimenten enthalten waren (d. h. ein anderer Barcode als NB01, NB05, NB09 oder NB10). Wir definierten diese als falsch zugeordnete Reads (Abbildung 2A).
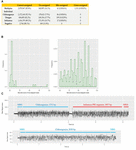
Abbildung 2. (A) Zusammenfassung der Anzahl und des Prozentsatzes der korrekt zugewiesenen, nicht zugewiesenen, falsch zugewiesenen und falsch zugewiesenen Reads in jedem Sequenzierungslauf. Nicht zugewiesen bezieht sich auf Reads, die von Albacore aufgrund eines Barcode-Scores von weniger als 60 keinem Bins zugewiesen werden können, falsch zugewiesen bezieht sich auf Reads, die Barcode-Bins zugewiesen wurden, die in diesem Experiment nicht enthalten sind, und falsch zugewiesen bezieht sich auf Reads, die den falschen Barcode-Bins zugewiesen wurden; (B) Verteilung der von Albacore gemeldeten Barcode-Scores für falsch zugewiesene Reads und falsch zugewiesene Reads in den Multiplex-Sequenzierungsdaten; (C) Vergleich des Rohsignals eines chimären und eines korrekt zugewiesenen Reads. Das Signal des chimären Reads weist ein Stallsignal und ein großes Spike-Signal in der Mitte des Reads auf.
Um eine mögliche Laborkontamination bei der Vorbereitung der Sequenzierbibliothek zu untersuchen, haben wir alle Reads aus jedem einzelnen Lauf gegen die genomischen Sequenzen aller drei Viren gemappt. Es wurde kein Read gefunden, der von einem Genom aus einer anderen Bibliothek stammte, was darauf schließen lässt, dass keine In-vitro-Kontamination vorliegt. Die Multiplex-Sequenzierungsbibliothek wurde durch Pooling der einzelnen, nicht kontaminierten Bibliotheken nach der Ligation von Barcode und Adapter hergestellt. Die Mapping-Ergebnisse zeigen jedoch, dass 1.311 (0,0543 %) Reads auf das falsche Zielgenom gemappt wurden, was darauf hindeutet, dass sie den falschen Barcode-Bins kreuzweise zugeordnet wurden (später als „kreuzweise zugeordnete Reads“ bezeichnet), obwohl die Multiplex-Sequenzierungsbibliothek mit einzelnen Bibliotheken gepoolt wurde und überhaupt keine kreuzweise zugeordneten Reads aufwies. Wir stellten die Hypothese auf, dass falsch zugewiesene und kreuzweise zugewiesene Reads auf einen niedrigen Barcode-Score zurückzuführen sind, und untersuchten die Barcode-Scores dieser Reads. Die meisten der falsch zugeordneten Reads hatten einen Barcode-Score <70, die falsch zugeordneten Reads hatten jedoch unterschiedlichere Scores, die von 60 bis fast 100 reichten (Abbildung 2B). Dieses Ergebnis deutet darauf hin, dass falsch zugewiesene und über Kreuz zugewiesene Reads aus unterschiedlichen Quellen stammen. Wir verglichen die kreuzzugeordneten Reads mit einer kleinen Datenbank, die die genomischen Sequenzen der drei in dieser Studie untersuchten Viren enthält, und zeigten, dass 1074/1311 (82 %) dieser Reads zu mehr als einem viralen Genom (1.047 Reads) oder zu verschiedenen Regionen innerhalb desselben Genoms (27 Reads) kreuzzugeordnet werden konnten, was darauf hindeutet, dass es sich um Chimären handelt. Um diese Beobachtung zu bestätigen, untersuchten wir die rohen Stromsignale einiger kreuzzugeordneter Reads im Vergleich zu denen der korrekt zugeordneten Reads (Abbildung 2C). Die Stromsignale eines korrekt zugeordneten Reads umfassen in der Regel: (i) ein Open-Pore-Signal mit hohem Strom, das die Zeit angibt, in der die Sequenzierpore von einem Adapter zu einem anderen wechselt, (ii) ein Stall-Signal, das sich auf die Zeitspanne bezieht, in der sich eine DNA-Sequenz in der Pore befindet, sich aber noch nicht bewegt, und (iii) die Signalspur der DNA-Sequenzierung. Im Gegensatz dazu weist ein chimärer Read ein stall-Signal und ein großes spike-Signal in der Mitte des Reads auf. Chimäre Reads können am Anfang und am Ende zwei verschiedene Barcode-Sequenzen aufweisen, was die Zuordnung zu einem Barcode-Bin verwirrt. Zusammengenommen zeigen diese Daten zwei Fehlerkategorien, die zu einer Kreuzprobenkontamination in unserem Datensatz beitragen: (i) chimäre Reads (die ∼80 % aller kreuzweise zugeordneten Reads ausmachen); (ii) Reads mit niedrigem Barcode-Score. Um die Qualität unseres endgültigen Datensatzes zu verbessern, untersuchten wir die Auswirkungen verschiedener Barcode-Demultiplexing-Ansätze, um kreuzzugeordnete Reads zu entfernen (Tabelle 3). Durch das Filtern der Reads, die einen internen Adapter besitzen, können 90 % der kreuzzugeordneten Reads entfernt werden, wobei 24 % der gesamten Reads verloren gehen. Wir haben auch ein strengeres Filterschema ausprobiert, das zwei Barcodes (jeweils einen am Anfang und am Ende des Reads) für eine Zuordnung erforderte. Bei diesem Ansatz wurden alle bis auf zwei kreuzweise zugeordnete Reads entfernt, aber 56 % der gesamten Reads gingen verloren.

TABLE 3. Entfernung von kreuzweise zugeordneten Reads und Verlust von Gesamtsequenzierungsdaten durch zwei Filterungsansätze mit Porechop.
Wir untersuchen auch das Ausmaß potenzieller chimärer Reads in den Sequenzierungsdaten. Für die einzelnen Sequenzierungsläufe von CHIKV, DENV und FLU-A zeigen die Mapping-Ergebnisse, dass 2,3, 3,0 bzw. 2,7 % der gemappten Reads ein zusätzliches Alignment besitzen und mindestens zweimal an dasselbe Genom angeglichen wurden (Tabelle 4). Wir betrachten sowohl die mit Barcode klassifizierten als auch die nicht klassifizierten Reads in den Multiplex-Sequenzierungsdaten. Die Ergebnisse zeigen, dass 2,0 % der gemappten Reads ein zusätzliches Alignment besitzen und mindestens zweimal an dasselbe Genom angeglichen wurden, während 0,052 % der gesamten Reads an mindestens zwei verschiedene Genome angeglichen wurden.

TABLE 4. Zusammenfassung der Anzahl und des Prozentsatzes der nicht-chimären, selbst-chimären und kreuz-chimären Reads in jedem Sequenzierungslauf.
Diskussion
Das Endziel unserer Forschung ist die Entwicklung eines diagnostischen Assays auf der Grundlage der Nanopore-Metagenom-Sequenzierung, der Tests für Infektionskrankheiten am Ort der Behandlung ermöglicht. Die Multiplex-Sequenzierung bietet die Möglichkeit, die Skalierbarkeit zu verbessern und die Kosten zu senken, allerdings kann eine Kreuzkontamination der Proben zu Fehlern in den Daten und einer falschen Interpretation der Ergebnisse führen.
In diesem Experiment haben wir saubere Bibliotheken gepoolt und eine Multiplex-MINION-Sequenzierung durchgeführt, um das Ausmaß und die Quelle der Kreuzkontamination zu untersuchen. Wir stellten fest, dass 0,056 % der gesamten Reads den falschen Barcode-Bins zugeordnet waren, was mit den Werten vergleichbar ist, die für Illumina-Sequenzierungsplattformen in verschiedenen Studien berichtet wurden (zwischen 0,06 und 0,25 %) (Nelson et al., 2014; D’Amore et al., 2016; Wright und Vetsigian, 2016). Unsere Ergebnisse zeigten, dass chimäre Reads die vorherrschende Quelle für Cross-Barcode-Zuordnungsfehler sind. Die kreuzweise zugeordneten chimären Reads in diesem Datensatz konnten nur während der Sequenzierung und nicht bei der Bibliotheksvorbereitung entstanden sein, da sie in den Sequenzierungsdaten der einzelnen Bibliotheken überhaupt nicht vorhanden waren und der einzige weitere Verarbeitungsschritt darin bestand, die endgültigen Sequenzierungsbibliotheken vor dem Laden zu mischen. Wir stellen die Hypothese auf, dass der aktuelle in Albacore implementierte Algorithmus die kurze Dissoziation zwischen DNA-Sequenzen, die gleichzeitig durch die Nanopore laufen, nicht erkennen kann, wodurch mehr als eine Sequenz in dieselbe Fast5-Datei verkettet wird.
Chimäre Reads wurden in MinION-Sequenzierungsdaten bereits in White et al. (2017) beobachtet. Durch Analysen der MinION-Sequenzierungsdaten von drei verschiedenen Interferon-Amplikonen fanden die Autoren heraus, dass 1,7 % der gemappten Reads Chimären waren. Unsere Ergebnisse tragen zu dem Wissen bei, dass Chimären in MinION-Sequenzierungsdaten häufig sind. Wir haben festgestellt, dass zwischen 2 und 3 % der gesamten Reads in drei Einzel- und einem Multiplex-Sequenzierdaten Chimären sind. Unsere Studie unterscheidet sich von früheren Arbeiten in den folgenden zwei Aspekten. Erstens liefern wir den direkten Beweis, dass chimäre Reads nach der Bibliotheksvorbereitung und während der Sequenzierung gebildet werden können; außerdem haben wir diese Chimären mit der Kreuzprobenkontamination bei der Multiplex-MinION-Sequenzierung in Verbindung gebracht, wie oben beschrieben. Andererseits ist unser Versuchsaufbau bei der Identifizierung potenzieller Chimären, die bei der Bibliotheksvorbereitung gebildet werden, eingeschränkt, insbesondere während des Adaptor-Ligationsschritts im Standard-Multiplex-Sequenzierungsprotokoll. Zweitens spiegeln unsere Ergebnisse den aktuellen Stand der MinION-Sequenzierung wider, da wir neuere und repräsentative ONT-Sequenzierungskits verwendet haben, darunter das Ligations-Sequenzierungskit 1D (SQK-LSK108) und das native Barcoding Kit 1D (EXP-93 NBD103). Die Nanopore-Sequenzierungstechnologie befindet sich in einer rasanten Entwicklung und wird in allen Bereichen verbessert. So wurden beispielsweise ein neueres DNA-Ligations-Sequenzierungskit (SQK-LSK109) und ein Kit für die direkte RNA-Sequenzierung (SQK-RNA001) auf den Markt gebracht; der in Albacore und Guppy Basecaller implementierte Basecalling-Algorithmus wurde verbessert. All diese Änderungen haben Auswirkungen auf das Ausmaß von Chimären in Nanopore-Sequenzierungsdaten und auf die Cross-Barcode-Kontamination während der Multiplex-Sequenzierung. Die Einschränkung dieser Studie war die geringe Anzahl von Experimenten. Weitere Arbeiten mit anderen Versuchsaufbauten würden unser Verständnis von Nanopore-Multiplex-Sequenzierungsdaten verbessern. Darüber hinaus ist es wichtig, die Beiträge potenzieller Faktoren zur Cross-Barcode-Kontamination zu untersuchen, was Aufschluss über bewährte Verfahren zur Analyse von Multiplex-Sequenzierungsdaten geben würde.
Zusammenfassend hat unsere Studie gezeigt, dass chimäre Reads die vorherrschende Quelle von Cross-Barcode-Zuordnungsfehlern bei der Multiplex-MinION-Sequenzierung sind. Sie unterstreicht die Notwendigkeit einer sorgfältigen Filterung von Multiplex-MINION-Sequenzierungsdaten vor der nachgeschalteten Analyse und den Kompromiss zwischen Sensitivität und Spezifität, der für die Barcode-Demultiplexing-Methoden gilt.
Beiträge der Autoren
SP, KL, SL und YX führten die MinION-Sequenzierung durch. YX analysierte die Daten. Alle Autoren konzipierten die Studie, beteiligten sich an der Interpretation der Ergebnisse und dem Verfassen des Manuskripts und lasen und genehmigten die endgültige Version dieses Manuskripts.
Finanzierung
Diese Arbeit wurde vom NIHR Oxford Biomedical Research Centre unterstützt.
Erklärung zu Interessenkonflikten
Die Autoren erklären, dass die Forschung in Abwesenheit jeglicher kommerzieller oder finanzieller Beziehungen durchgeführt wurde, die als potenzieller Interessenkonflikt ausgelegt werden könnten.
Dankesworte
Wir danken Dr. Anthony Marriott (Public Health England) für die Bereitstellung von Frettchen-Nasenaspiraten.