- Introduction
- Matériel et méthodes
- Préparation des échantillons
- Préparation des librairies MinION et séquençage
- Analyse génomique
- Résultats
- Données de séquençage MinION et assemblage de génomes viraux
- Etendue et source de la contamination inter-échantillons
- Discussion
- Contributions des auteurs
- Financement
- Déclaration de conflit d’intérêts
- Remerciements
- Matériel supplémentaire
- Notes de bas de page
Introduction
Le séquençage métagénomique a le potentiel de permettre une identification non biaisée des agents pathogènes à partir d’un échantillon clinique. Il est prometteur de servir de test unique et universel pour le diagnostic des maladies infectieuses directement à partir d’échantillons sans avoir besoin de connaissances a priori (Bibby, 2013 ; Miller et al., 2013 ; Schlaberg et al., 2017). Outre l’identification des espèces pathogènes, les données de séquences métagénomiques larges et profondes pourraient fournir des informations pertinentes pour déterminer le traitement et le pronostic, détecter les épidémies et suivre l’épidémiologie des infections (Greninger et al., 2010 ; Yang et al., 2011 ; Qin et al., 2012 ; Loman et al., 2013). Les plateformes de séquençage de nouvelle génération (NGS) peuvent produire un débit massif de données à un coût modeste, cependant, son application dans le diagnostic clinique et la santé publique a été limitée par la complexité, la lenteur et l’investissement en capital.
Le MinION est un séquenceur de génome monomoléculaire en temps réel, de la taille d’une paume, développé par Oxford Nanopore Technologies (ONT). La taille compacte du MinION et sa nature en temps réel pourraient faciliter l’application du séquençage métagénomique dans les tests au point de service pour les maladies infectieuses, comme le démontrent plusieurs études de preuve de concept, notamment l’identification du Chikungunya (CHIKV), de l’Ebola (EBOV) et du virus de l’hépatite C (HCV) à partir d’échantillons de sang clinique humain sans enrichissement de la cible (Greninger et al, 2015), et la détection d’agents pathogènes bactériens à partir d’échantillons d’urine (Schmidt et al., 2016) et d’échantillons respiratoires, sans culture préalable (Pendleton et al., 2017).
Le débit de données de MinION a considérablement augmenté depuis sa sortie en 2015, chaque cellule à flux consommable générant désormais jusqu’à 10-20 Go de données de séquences d’ADN. Cela permet aux utilisateurs de faire un usage plus efficace de la cellule à flux (et de réduire les coûts) en multiplexant plusieurs échantillons en un seul passage de séquençage. ONT a développé des jeux de codes-barres sans PCR qui permettent de multiplexer jusqu’à 12 échantillons.
La détection du virus de la grippe A dans plusieurs échantillons respiratoires pourrait être l’une des utilisations diagnostiques d’un test de séquençage MinION multiplexé. Cependant, lors du séquençage direct d’échantillons avec une large gamme potentielle de titres viraux, il est important d’être conscient du potentiel de contamination croisée des échantillons, à la fois pendant la préparation de la bibliothèque et l’étape de démultiplexage bioinformatique des codes-barres après le séquençage. Nous présentons ici un ensemble unique de données de séquençage MinION et les résultats d’une enquête sur l’étendue et la source de la contamination croisée des codes-barres dans le séquençage multiplex.
Matériel et méthodes
Nous avons utilisé un échantillon de lavage nasal de furet infecté par le virus de la grippe A comme exemplaire et avons également dopé séparément deux aliquotes d’échantillons de lavage nasal négatif de furet non infecté (stocks préexistants non utilisés provenant d’une étude non liée) avec des virus de la dengue et du chikungunya. Aucun de ces virus n’est pertinent pour le diagnostic clinique dans les échantillons respiratoires, mais ils servent ici de marqueurs clairs et distincts pour l’évaluation de la contamination croisée des échantillons. Les bibliothèques de séquençage de chaque échantillon ont été préparées en parallèle, ainsi qu’un contrôle négatif de lavage nasal, avec un code-barres, et séquencées individuellement. Nous avons ensuite regroupé une aliquote des bibliothèques de séquençage et effectué un séquençage MinION multiplex. Les lectures des quatre passages individuels (appelés » CHIKV « , » DENV « , » FLU-A » et » Négatif « ) et du passage multiplex (appelé » Multiplexé « ) ont ensuite été analysées pour étudier l’étendue et la source de la contamination croisée des échantillons.
Préparation des échantillons
La licence du projet a été examinée par l’AWERB (Animal Welfare and Ethics Review Board) local et a ensuite été accordée par le Home Office. L’ARN a été extrait, à l’aide du kit d’ARN viral QIAamp (Qiagen) selon les instructions du fabricant, du lavage nasal de furet contenant le virus de la grippe A (H1N1) (A/California/04/2009) et d’un pool d’échantillons de lavage nasal négatifs. Des aliquotes d’extraits d’échantillons négatifs ont été dopés avec de l’ARN viral de dengue (DENV) (souche TC861HA, GenBank : MF576311) ou de CHIKV (souche S27, GenBank : MF580946.1) provenant de la National collection of Pathogenic Viruses1. Les échantillons ont été traités à la DNase à l’aide de TURBO DNase (Thermo Fisher Scientific, Waltham, MA, États-Unis) et purifiés à l’aide du kit RNA Clean & ConcentratorTM-5 (Zymo Research). L’ADNc a été préparé et amplifié à l’aide de méthodes d’amplification Sequence-Independent-Single-Primer-Amplification (Greninger et al., 2015) modifiées comme décrit précédemment (Atkinson et al., 2016). L’ADNc amplifié a été quantifié à l’aide du kit de test Qubit dsDNA HS (Thermo Fisher Scientific, Waltham, MA, États-Unis), et 1 μg a été utilisé comme entrée pour chaque préparation de bibliothèque MinION, à l’exception du contrôle négatif où l’échantillon entier (32 ng) a été utilisé.
Préparation des librairies MinION et séquençage
Le kit de séquençage par ligature 1D (SQK-LSK108) et le kit de code-barres natif 1D (EXP-NBD103) ont été utilisés conformément aux protocoles standard de l’ONT, à l’exception du fait qu’un seul code-barres a été inclus dans chacune des quatre préparations de librairies. Chaque bibliothèque a été exécutée sur une cellule d’écoulement individuelle et une cinquième bibliothèque groupée a été réalisée en combinant les quatre bibliothèques à code-barres individuelles. Les librairies ont été séquencées sur des cellules à flux R9.4. Le plan de l’étude est présenté à la figure 1.
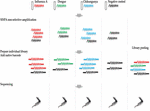
FIGURE 1. Vue d’ensemble du plan d’étude. L’ARN a été extrait de quatre échantillons, dont un échantillon de lavage nasal de furet infecté par le virus de la grippe A, deux échantillons de lavage nasal de furet négatifs dopés par les virus de la dengue et du chikungunya, et un contrôle de lavage nasal de furet négatif. L’ADNc a été préparé et amplifié à l’aide de méthodes d’amplification Sequence-Independent-Single-Primer-Amplification. Les bibliothèques de séquençage de chaque échantillon ont été préparées en parallèle, munies d’un code-barres et séquencées sur des cellules d’écoulement individuelles. Le séquençage multiplex a également été réalisé en regroupant les quatre bibliothèques individuelles. Les lectures des quatre parcours individuels et du parcours multiplex ont été analysées afin d’évaluer l’étendue et la source de la contamination par code-barres croisé lors du séquençage multiplex.
Analyse génomique
Les lectures ont été appelées en base à l’aide d’Albacore v2.1.7 (ONT) avec démultiplexage du code-barres. Les lectures de chaque cycle de séquençage ont été mappées aux séquences génomiques de chaque virus en utilisant Minimap2 (Li, 2018). Le nombre de lectures mappées à la référence a été compté en utilisant Pysam2. L’assemblage de novo a été effectué à l’aide de Canu v1.7 (Koren et al., 2017), et l’ébauche de génome résultante a été polie à l’aide de Nanopolish (Mongan et al., 2015) avec les données de niveau de signal.
Pour permettre un démultiplexage rigoureux des codes-barres des données de séquençage MinION multiplex, nous avons effectué deux séries d’analyses à l’aide de Porechop (v0.2.23). La présence d’une séquence adaptatrice au milieu d’une lecture est une signature de chimère. Nous avons utilisé Porechop pour examiner chaque lecture et celles dont la région centrale partage >75% d’identité avec la séquence adaptatrice ont été identifiées comme des lectures chimériques. Dans Porechop, nous avons défini l’option « -middle_threshold » et choisi un seuil de 75. Dans le deuxième tour, nous avons utilisé Porechop pour rechercher la séquence du code-barres au début et à la fin d’une lecture ; les lectures ont été assignées seulement si le même code-barres a été trouvé aux deux extrémités. Nous avons défini l’option « -require_two_barcodes » dans Porechop et fixé le seuil du score du code-barres à 70. Pour trouver la signature potentielle de lectures chimériques, nous avons examiné les signaux de courant de lecture stockés dans le fichier FAST5 par le séquenceur MinION. Les signaux de courant ont été extraits en utilisant ONT fast5 API4 et tracés en utilisant ggplot2 implémenté dans R5 pour une comparaison des lectures chimériques et non chimériques.
Résultats
Données de séquençage MinION et assemblage de génomes viraux
Le débit de chaque cycle de séquençage MinION a varié en raison des différences de temps d’exécution. Un nombre maximal de ∼2,4 M de lectures a été atteint par le run de séquençage multiplexé et le run individuel CHIKV, en raison de temps de fonctionnement plus longs (tableau supplémentaire S1). Les lectures provenant du virus piqué représentaient 96 % des données des cycles de séquençage individuels du CHIKV et du DENV, et 78 % pour l’échantillon FLU-A (tableau 1). Le pourcentage de lectures virales au sein de chaque échantillon à code-barres dans les données de séquençage multiplexé est proche de celui des données des échantillons analysés individuellement (tableau 2). Chaque génome viral avait une profondeur de couverture moyenne ultra-haute (>8 000) dans les données de séquençage individuel et multiplexé, et l’assemblage de novo a permis de récupérer des génomes presque complets pour les trois virus avec des identités de 99.9% d’identité par rapport à la référence GenBank.

TABLE 1. Résumé des résultats de cartographie et d’assemblage de novo pour les données issues du séquençage MinION de bibliothèques individuelles.

Tableau 2. Résumé des résultats de cartographie et d’assemblage de novo pour les données issues du séquençage MinION multiplex.
Etendue et source de la contamination inter-échantillons
Chaque échantillon a reçu un code-barres et a été séquencé à la fois individuellement et en multiplex, ce qui nous a permis d’examiner la performance du démultiplexage du code-barres du germon. Dans les données des échantillons séquencés individuellement, nous nous attendrions à ce qu’un seul code-barres natif soit présent. Pour les séries de séquençage individuelles de CHIKV (code-barres NB01), DENV (NB09) et FLU-A (NB10), nous avons constaté que 86, 109 et 17 lectures, respectivement, ont été attribuées à des cases de codes-barres dont on ne s’attendait pas à ce qu’ils soient présents dans la bibliothèque (représentant 0,0036, 0,0129 et 0,001 % du total des lectures). Dans les données de séquençage multiplex, 41 lectures (0,0016 %) ont été attribuées à des codes-barres non inclus dans les expériences (c’est-à-dire un code-barres autre que NB01, NB05, NB09 ou NB10). Nous les avons définis comme des lectures mal attribuées (figure 2A).
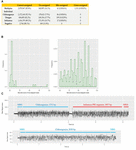
FIGURE 2. (A) résumé du nombre et du pourcentage de lectures correctement attribuées, non attribuées, mal attribuées et attribuées de manière croisée dans chaque cycle de séquençage. Les lectures non assignées sont celles qui ne peuvent être assignées à aucune case par Albacore en raison d’un score de code-barres inférieur à 60, les lectures mal assignées sont celles qui ont été assignées à des cases de code-barres non incluses dans cette expérience, et les assignations croisées sont celles qui ont été assignées à des cases de code-barres incorrectes ; (B) distribution des scores de code-barres signalés par Albacore pour les lectures mal assignées et les lectures assignées de manière croisée dans les données de séquençage multiplex ; (C) comparaison du signal brut d’une lecture chimérique et d’une lecture correctement assignée. Le signal d’une lecture chimérique possède un signal de décrochage et un énorme signal de pointe au milieu de la lecture.
Pour examiner la contamination potentielle du laboratoire dans la préparation de la bibliothèque de séquençage, nous avons cartographié toutes les lectures de chaque passage individuel par rapport aux séquences génomiques des trois virus. Aucune lecture n’a été trouvée comme provenant d’un génome préparé dans une bibliothèque différente, ce qui suggère l’absence de contamination in vitro. La bibliothèque de séquençage multiplex a été préparée en regroupant les bibliothèques individuelles non contaminées après la ligature du code-barres et de l’adaptateur. Cependant, les résultats de cartographie montrent que 1 311 (0,0543 %) lectures ont été cartographiées sur le mauvais génome cible, ce qui implique qu’elles ont été affectées aux mauvaises cases de code-barres (appelées par la suite « lectures affectées de manière croisée »), malgré le fait que la bibliothèque de séquençage multiplexée ait été regroupée avec les bibliothèques individuelles ne montrant aucune lecture affectée de manière croisée. Nous avons émis l’hypothèse que les lectures mal attribuées et attribuées de manière croisée étaient dues à un score de code-barres faible, et nous avons étudié les scores de code-barres de ces lectures. La plupart des lectures mal attribuées avaient un score de code-barres <70, cependant, les lectures attribuées de manière croisée avaient des scores plus diversifiés allant de 60 à près de 100 (Figure 2B). Ce résultat suggère que les lectures mal attribuées et les lectures attribuées de manière croisée proviennent de sources différentes. Nous avons comparé les lectures croisées à une petite base de données comprenant les séquences génomiques des trois virus inclus dans cette étude et avons démontré que 1074/1311 (82%) de ces lectures pouvaient être alignées sur plus d’un génome viral (1047 lectures) ou alignées sur des régions distinctes du même génome (27 lectures), ce qui suggère qu’il s’agit de chimères. Pour confirmer cette observation, nous avons étudié les signaux de courant bruts de quelques lectures à alignement croisé par rapport à ceux des lectures correctement assignées (Figure 2C). Les signaux de courant d’une lecture correctement assignée comprennent généralement : (i) un signal de pore ouvert de courant élevé représentant le temps pendant lequel le pore de séquençage passe d’un adaptateur à un autre, (ii) un signal de décrochage, se référant à la période de temps pendant laquelle une séquence d’ADN est dans le pore mais n’a pas encore bougé, et (iii) la trace du signal de séquençage de l’ADN. En revanche, une lecture chimérique possède un signal de décrochage et un énorme signal de pointe au milieu de la lecture. Les lectures chimériques peuvent posséder deux séquences de code-barres différentes au début et à la fin, ce qui rend confuse l’attribution d’une case de code-barres. Prises ensemble, ces données démontrent deux catégories d’erreurs qui contribuent à la contamination croisée des échantillons dans notre ensemble de données : (i) les lectures chimériques (qui représentent ∼80 % de toutes les lectures à affectation croisée) ; (ii) les lectures avec un faible score de code-barres. Afin d’améliorer la qualité de notre jeu de données final, nous avons exploré l’impact de différentes approches de démultiplexage des codes-barres pour éliminer les lectures à affectation croisée (tableau 3). Le filtrage des lectures qui possèdent un adaptateur interne peut éliminer 90% des lectures à affectation croisée et perdre 24% des lectures totales. Nous avons également essayé un schéma de filtrage plus rigoureux qui exigeait deux codes-barres (un au début et à la fin de la lecture) pour effectuer une assignation. Cette approche a éliminé toutes les lectures à affectation croisée sauf deux, mais a perdu 56 % des lectures totales.

TABLE 3. Suppression des lectures affectées par croisement et perte de données de séquençage totales par deux approches de filtrage utilisant Porechop.
Nous étudions également l’étendue des lectures chimériques potentielles dans les données de séquençage. Pour les séries de séquençage individuelles CHIKV, DENV et FLU-A, les résultats de cartographie montrent que 2,3, 3,0 et 2,7 % des lectures cartographiées, respectivement, possèdent un alignement supplémentaire et s’alignent au moins deux fois sur le même génome (tableau 4). Nous considérons à la fois les lectures classées et non classées par code-barres dans les données de séquençage multiplex. Les résultats montrent que 2,0% des lectures mappées possèdent un alignement supplémentaire et se sont alignées au moins deux fois sur le même génome, tandis que 0,052% des lectures totales ont été alignées sur au moins deux génomes distincts.

TABLE 4. Résumé du nombre et du pourcentage de lectures non-chimériques, auto-chimériques et cross-chimériques dans chaque cycle de séquençage.
Discussion
L’objectif ultime de notre recherche est de développer un test de diagnostic basé sur le séquençage métagénomique nanopore qui permet un test au point de service pour les maladies infectieuses. Le séquençage multiplex offre la possibilité d’améliorer l’évolutivité et de réduire les coûts, cependant, la contamination croisée des échantillons peut entraîner des erreurs dans les données et une fausse interprétation des résultats.
Dans cette expérience, nous avons regroupé des bibliothèques propres et effectué un séquençage MinION multiplex afin d’étudier l’étendue et la source de la contamination croisée des codes-barres. Nous avons identifié que 0,056 % des lectures totales étaient affectées de manière croisée aux bacs de code-barres incorrects, ce qui est comparable à ceux rapportés pour les plateformes de séquençage Illumina par différentes études (entre 0,06 et 0,25 %) (Nelson et al., 2014 ; D’Amore et al., 2016 ; Wright et Vetsigian, 2016). Nos résultats ont montré que les lectures chimériques sont la source prédominante des erreurs d’assignation de codes-barres croisés. Les lectures chimériques à affectation croisée dans cet ensemble de données n’ont pu se former que pendant le séquençage plutôt que pendant la préparation des bibliothèques, car elles étaient totalement absentes des données de séquençage des bibliothèques individuelles, et la seule étape de traitement supplémentaire consistait à mélanger les bibliothèques de séquençage finales avant le chargement. Nous émettons l’hypothèse que l’algorithme actuel implémenté dans Albacore ne peut pas reconnaître la courte dissociation entre les séquences d’ADN qui passent simultanément à travers le nanopore, concaténant ainsi plus d’une séquence dans le même fichier Fast5.
Des lectures chimériques ont été observées dans les données de séquençage MinION avant dans White et al. (2017). Grâce aux analyses des données de séquençage MinION de trois amplicons d’interféron différents, les auteurs ont constaté que 1,7 % des lectures mappées étaient des chimères. Nos résultats s’ajoutent aux connaissances soutenant que les chimères sont courantes dans les données de séquençage MinION. Nous avons identifié entre 2 et 3% des lectures totales dans trois données de séquençage individuel et une donnée de séquençage multiplex sont des chimères. Notre étude se distingue des travaux précédents par les deux aspects suivants. Tout d’abord, nous fournissons des preuves directes que des lectures chimériques peuvent être formées après la préparation de la bibliothèque et pendant le séquençage ; nous avons ensuite lié ces chimères à la contamination croisée des échantillons dans le séquençage MinION multiplex, comme indiqué ci-dessus. D’autre part, la configuration de notre expérience est limitée dans l’identification des chimères potentielles formées lors de la préparation de la bibliothèque, en particulier pendant l’étape de ligature de l’adaptateur dans le protocole standard de séquençage multiplex. Deuxièmement, nos résultats reflètent l’état actuel du séquençage MinION car nous avons utilisé le kit de séquençage ONT le plus récent et le plus représentatif, notamment le kit de séquençage par ligature 1D (SQK-LSK108) et le kit de codage à barres natif 1D (EXP-93 NBD103). La technologie de séquençage Nanopore est en plein développement et des améliorations sont apportées dans tous les domaines. Par exemple, un nouveau kit de séquençage par ligature de l’ADN (SQK-LSK109) et un kit de séquençage direct de l’ARN (SQK-RNA001) ont été mis sur le marché ; l’algorithme d’appel de base mis en œuvre dans les séquenceurs Albacore et Guppy a été amélioré. Tous ces changements ont un effet sur l’étendue des chimères dans les données de séquençage Nanopore et sur la contamination des codes à barres croisés pendant le séquençage multiplex. La limite de cette étude était le petit nombre d’expériences, des travaux supplémentaires utilisant différentes configurations d’expériences permettraient d’améliorer notre compréhension des données de séquençage multiplex de Nanopore. En outre, il est important d’étudier les contributions des facteurs potentiels à la contamination par code-barres croisé, ce qui éclairerait les meilleures pratiques d’analyse des données de séquençage multiplex.
En résumé, notre étude a démontré que les lectures chimériques sont la source prédominante d’erreurs d’attribution de codes-barres croisés dans le séquençage MinION multiplex. Elle met en évidence la nécessité de filtrer soigneusement les données de séquençage MinION multiplex avant l’analyse en aval, et le compromis entre sensibilité et spécificité qui s’applique aux méthodes de démultiplexage des codes-barres.
Contributions des auteurs
SP, KL, SL et YX ont réalisé le séquençage MinION. YX a analysé les données. Tous les auteurs ont conçu l’étude, participé à l’interprétation des résultats et à la rédaction du manuscrit, et lu et approuvé la version finale de ce manuscrit.
Financement
Ce travail a été soutenu par le NIHR Oxford Biomedical Research Centre.
Déclaration de conflit d’intérêts
Les auteurs déclarent que la recherche a été menée en l’absence de toute relation commerciale ou financière qui pourrait être interprétée comme un conflit d’intérêts potentiel.
Remerciements
Nous tenons à remercier le Dr Anthony Marriott (Public Health England) pour avoir fourni des aspirations nasales de furet.
Matériel supplémentaire
Notes de bas de page
.