Plusieurs des méthodes de visualisation et d’interprétation des données d’expression génique peuvent être utilisées à la fois pour les expériences microarray et RNA-seq. Certaines des méthodes les plus courantes sont abordées ci-dessous.
Cartes thermiques et clustering
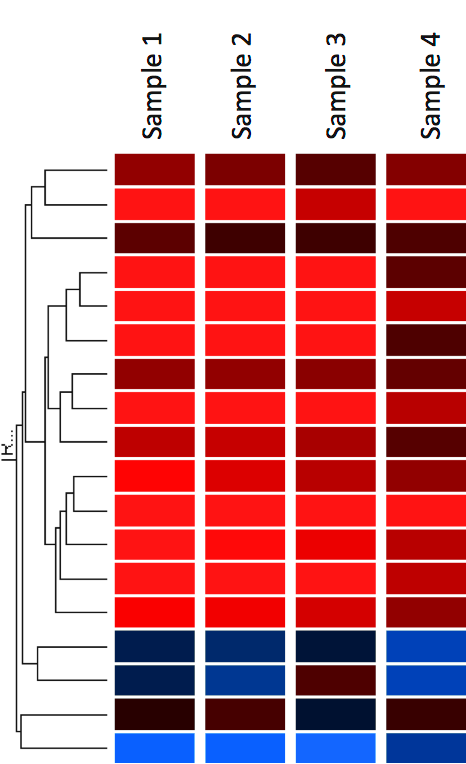
Une méthode courante de visualisation des données d’expression génique consiste à les afficher sous forme de carte thermique (figure 12). La carte thermique peut également être combinée avec des méthodes de clustering qui regroupent les gènes et/ou les échantillons sur la base de la similarité de leur schéma d’expression génique. Cela peut être utile pour identifier les gènes qui sont couramment régulés, ou les signatures biologiques associées à une condition particulière (par exemple une maladie ou une condition environnementale) (4).
Dans les cartes thermiques, les données sont affichées dans une grille où chaque ligne représente un gène et chaque colonne un échantillon. La couleur et l’intensité des cases sont utilisées pour représenter les changements (et non les valeurs absolues) de l’expression des gènes. Dans l’exemple ci-dessous, le rouge représente les gènes régulés à la hausse et le bleu les gènes régulés à la baisse. Le noir représente une expression inchangée.
Analyse d’enrichissement des ensembles de gènes et analyse des voies
Une approche courante pour interpréter les données d’expression génique est l’analyse d’enrichissement des ensembles de gènes basée sur l’annotation fonctionnelle des gènes différentiellement exprimés (Figure 13). Cette méthode est utile pour savoir si les gènes différentiellement exprimés sont associés à un certain processus biologique ou à une fonction moléculaire.
La Gene Ontology, qui contient une annotation standardisée des produits des gènes, est couramment utilisée à cette fin. Elle fonctionne en comparant la fréquence des annotations individuelles dans la liste de gènes (par exemple, les gènes différentiellement exprimés) avec une liste de référence (généralement tous les gènes de la micro-puce ou du génome). L’enrichissement des voies biologiques fournies par KEGG, Ingenuity, Reactome ou WikiPathways peut être réalisé de manière similaire (12,13).
Les outils populaires pour l’enrichissement des ensembles de gènes et l’analyse des voies biologiques comprennent :
- DAVID (outil en ligne gratuit)
- GSEA (gratuit)
- Ingenuity (licence requise)
- Ractome (gratuit)

Analyse de réseau
L’analyse de réseau est complémentaire à l’analyse des voies et peut être utilisée pour montrer comment les composants clés de différentes voies interagissent. Cela peut être utile pour identifier les événements régulateurs qui influencent de multiples processus et voies biologiques (12,13).
L’analyse des réseaux est un complément à l’analyse des voies.