Outcome des fœtus présentant un Cavum Septi Pellucidi anormal : Expérience d’un centre tertiaire
George Lucian Zorilaa, Stefania Tudorachea, Elena-Madalina Barbua, Maria-Cristina Comanescua, Razvan-Grigoras Capitanescua, Marius-Cristian Marinasb, Maria Floreaa, Nicolae Cerneaa, Dominic Gabriel Iliescua
aUnité de diagnostic prénatal, département d’obstétrique et de gynécologie, hôpital départemental des urgences universitaires, Craiova, Roumanie
bDépartement d’anatomie, université de médecine et de pharmacie de Craiova, Roumanie
cAuteur correspondant : Stefania Tudorache, Prenatal Diagnosis Unit, Department of Obstetrics and Gynecology, University Emergency County Hospital, Craiova, Romania
Manuscript accepted for publication December 28, 2016
Short title: Fetuses With Abnormal Cavum Septi Pellucidi
doi: https://doi.org/10.14740/jcgo423w
- Abstract
- Introduction
- Methods
- Results
- Discussion
| Abstract | ▴Top |
Background: Cavum septi pellucidi (CSP) is easily evaluated in the second and third trimester of the pregnancy. Cette structure est une caractéristique importante des plans standards utilisés pour l’évaluation morphologique de routine de la tête et du système nerveux central (SNC) du fœtus : plan trans-thalamique et plan trans-ventriculaire. La description standard du CSP est une boîte rectangulaire anéchogène entre deux lignes hyperéchogènes représentées par le septum pellucidum. Les aspects pathologiques sont principalement représentés par l’absence de son apparence normale, qui est associée à des malformations graves du SNC de la ligne médiane du cerveau comme l’agénésie du corps calleux, l’hydranencéphalie, la porencéphalie, la schizencéphalie, l’holoprosencéphalie, la syntélencéphalie ou l’hydrocéphalie chronique grave. D’autres problèmes tels que des dimensions accrues ou réduites de la CSP ne sont pas considérés comme significatifs s’ils sont trouvés isolément, bien que l’investigation génétique fœtale soit suggérée par certains auteurs. L’objectif de l’étude était d’évaluer l’issue des fœtus présentant une CSP anormale détectée par échographie au cours des 5 dernières années, dans l’unité de diagnostic prénatal (PDU) de notre centre tertiaire.
Méthodes : Nous avons effectué une revue rétrospective des cas de CSP anormale évalués dans notre unité tertiaire, diagnostiqués entre janvier 2012 et novembre 2016. L’anatomie fœtale a été évaluée dans tous les cas en suivant les recommandations des directives internationales. Dans les cas de CSP anormale, un neurosonogramme fœtal et un scanner d’anomalie fœtale étendu ont été réalisés et une amniocentèse a été proposée pour identifier les troubles génétiques.
Résultats : Un total de 7 520 cas ont été examinés à des fins morphologiques et une CSP anormale a été trouvée dans 36 cas. L’absence de CSP était l’observation initiale qui a déclenché des investigations plus poussées et le diagnostic dans les cas d’agénésie du corps calleux (ACC) (sept cas) et de dysplasie septo-optique (deux cas). Dans l’hydranencéphalie ou l’hydrocéphalie sévère, la porencéphalie, la schizencéphalie et l’holoprosencéphalie, l’aspect fortement malformé du cerveau est évident, et l’absence de CSP n’est qu’une observation, avec moins d’importance diagnostique et d’implications cliniques. L’ACC partiel ou total était principalement associé à l’absence de CSP, car le développement des deux structures est fusionné. Près de la moitié des cas de CSP anormale (16/36) étaient associés à des troubles génétiques, la plupart d’entre eux avec un caryotype anormal et tous étaient associés à l’absence de CSP. L’élargissement persistant du CSP (4/36 cas) et l’aspect hyperéchogène du CSP (1/36) n’étaient pas associés à d’autres anomalies structurelles ou génétiques et le développement neuromoteur postnatal était normal.
Conclusions : L’évaluation du CSP est obligatoire et les aspects normaux suggèrent un développement normal du mésencéphale. Les tests génétiques devraient être proposés en particulier pour les cas d’absence de CSP en raison de l’incidence élevée des troubles chromosomiques. Comme l’absence de CSP est associée à de graves troubles structurels ou génétiques, sa visualisation dans la seconde moitié de la grossesse est obligatoire pour toute analyse d’anomalie. Les variations d’agrandissement et d’échogénicité de la CSP associent une issue néonatale favorable. Cependant, un suivi à long terme est recommandé chez les nouveau-nés et les nourrissons apparemment normaux, car ils peuvent développer un comportement psychologique anormal dernièrement.
Mots-clés : Cavum septi pellucidi ; Corpus callosum ; échographie ; neurosonogramme ; diagnostic prénatal ; Système nerveux central
| Introduction | ▴Top. |
Le cavum septi pellucidi (CSP) est une structure importante, développée conjointement avec le corps calleux (CC) à partir de 10 semaines d’âge de gestation (AG) jusqu’à 18 semaines d’AG. Elle est située au milieu du cerveau, au-dessus du fornix, entre les deux parois médianes des ventricules latéraux et sous le CC. Cette structure se poursuit postérieurement avec le cavum vergae et la limite entre ces deux est un plan entre les foramens Monroe. La plupart des auteurs utilisent le terme CSP pour « CSP et Vergae ». Nous nous conformons à ce terme et utilisons « CSP » pour les deux structures. Le septi pellucidi sert de station relais importante ; ses connexions de fibres anatomiques et fonctionnelles les plus importantes sont avec l’hippocampe et l’hypothalamus.
La CSP est systématiquement imagée après 18 semaines de gestation sur les trois vues obligatoires de la tête fœtale obtenues lors de l’échographie fœtale. Plus précisément, en plus des images des ventricules et de la fosse postérieure, une vue axiale au niveau des deux thalamis permet d’obtenir le diamètre bipariétal et le CSP . Une question importante pour le diagnostic de la CSP anormale est de ne pas confondre le fornix avec la CSP, en raison de leur proximité et de leur apparence relativement similaire. La CSP normale doit apparaître comme une boîte sombre due au liquide contenu dans le cavum, entourée de deux lignes blanches sur les deux côtés latéraux, représentées par le septum pellucidum. Au niveau du fornix, la boîte est divisée par une troisième ligne sagittale. Cette différenciation est d’une importance majeure, car le développement du fornix n’est pas lié au CC .
Les aspects anormaux du CSP comprennent son absence, son élargissement ou son échogénicité. L’élargissement normal de la CSP a été normalisé entre 2 et 4,7 mm (± 2 SD) à 19 – 20 semaines d’AG et jusqu’à 9 mm comme limite supérieure à 38 semaines, en considérant 2 SD également . L’élargissement de la CSP a été associé à l’hydrocéphalie, aux translocations chromosomiques et au retard de croissance. Lorsqu’il s’agit d’une anomalie échographique isolée, l’issue fœtale est favorable, bien qu’une évaluation génétique fœtale doive être proposée. L’évolution normale du CSP après la naissance est la fermeture du cavum vergae au CSP, et seulement 15% de cet espace est visible à 6 mois après la naissance. Une CSP élargie de plus de 1 cm après la naissance, et la persistance d’une CSP après la petite enfance ont été décrites comme des « marqueurs subtils de dysgénésie cérébrale », possiblement associés à des troubles neuropsychiatriques, en particulier la schizophrénie. Ainsi, le suivi post-partum des cas présentant une hypertrophie prénatale de la CSP est important .
L’absence de CSP aux deuxième et troisième trimestres a été associée à une ACC, une dysplasie septo-optique, une hydranencéphalie, une porencéphalie, une schizencéphalie, une holoprosencéphalie, une syntélencéphalie ou une hydrocéphalie chronique sévère .
Les grandes études de la population prénatale concernant les associations morpho-génétiques des CSP anormaux et leurs taux sont utiles pour le conseil parental prénatal. Étant donné la faible prévalence de l’anomalie et son hétérogénéité, un grand nombre de cas est nécessaire. Notre série est l’une des plus importantes de la littérature, et le spectre de nos données offre la possibilité d’être incluse dans une méta-analyse plus large. Notre objectif était de rapporter le résultat des cas présentant tout aspect anormal de la CSP qui ne respecte pas les critères de normalité, par exemple une échogénicité anormale.
| Méthodes | ▴Top |
Les caractéristiques prénatales ont été analysées rétrospectivement à partir de la base de données échographiques, sur une période de 5 ans, entre janvier 2012 et novembre 2016. Les cas ont été examinés dans un centre tertiaire – l’unité de diagnostic prénatal (PDU) de l’hôpital universitaire d’urgence du comté, Craiova, Roumanie. L’anatomie fœtale a été évaluée dans tous les cas en suivant les recommandations des directives internationales . Les examens ont été réalisés par des obstétriciens spécialisés en médecine fœtale, ayant au moins 2 ans d’expérience en US prénatale. Les cas présentant des anomalies suspectées qui n’ont pu être confirmées lors de l’examen initial ont été invités à être réévalués. Lorsque des anomalies étaient suspectées lors de l’examen US, deux examinateurs expérimentés ont étudié le fœtus pour confirmer les anomalies. Dans les cas de CSP anormaux, un neurosonogramme fœtal et un examen étendu des anomalies fœtales ont été réalisés et une amniocentèse a été proposée pour identifier les troubles génétiques. Des échographies fœtales 2D et 3D ont été réalisées dans tous les cas présentant des anomalies du SNC (machines US Voluson 730 Pro, GE Medical Systems). Le post-traitement des volumes acquis était destiné à une meilleure visualisation des anomalies ou des caractéristiques associées. Après confirmation, des conseils et une prise en charge appropriés ont été fournis par une équipe interdisciplinaire (obstétricien, généticien, néonatologiste – pédiatre, chirurgien pédiatrique).
| Résultats | ▴Top. |
Durant l’intervalle de temps, nous avons examiné 7 520 grossesses à des fins morphologiques, des malformations du SNC ont été trouvées dans 257 cas, et la principale anomalie était la ventriculomégalie (Fig. 1). Une CSP anormale a été trouvée dans 36 cas comme suit : l’absence de CSP a été trouvée dans 31 cas (86,1% des anomalies de CSP), une CSP élargie a été mesurée dans quatre cas (11.1%) et une CSP hyperéchogène a été notée dans un cas (2,7%).
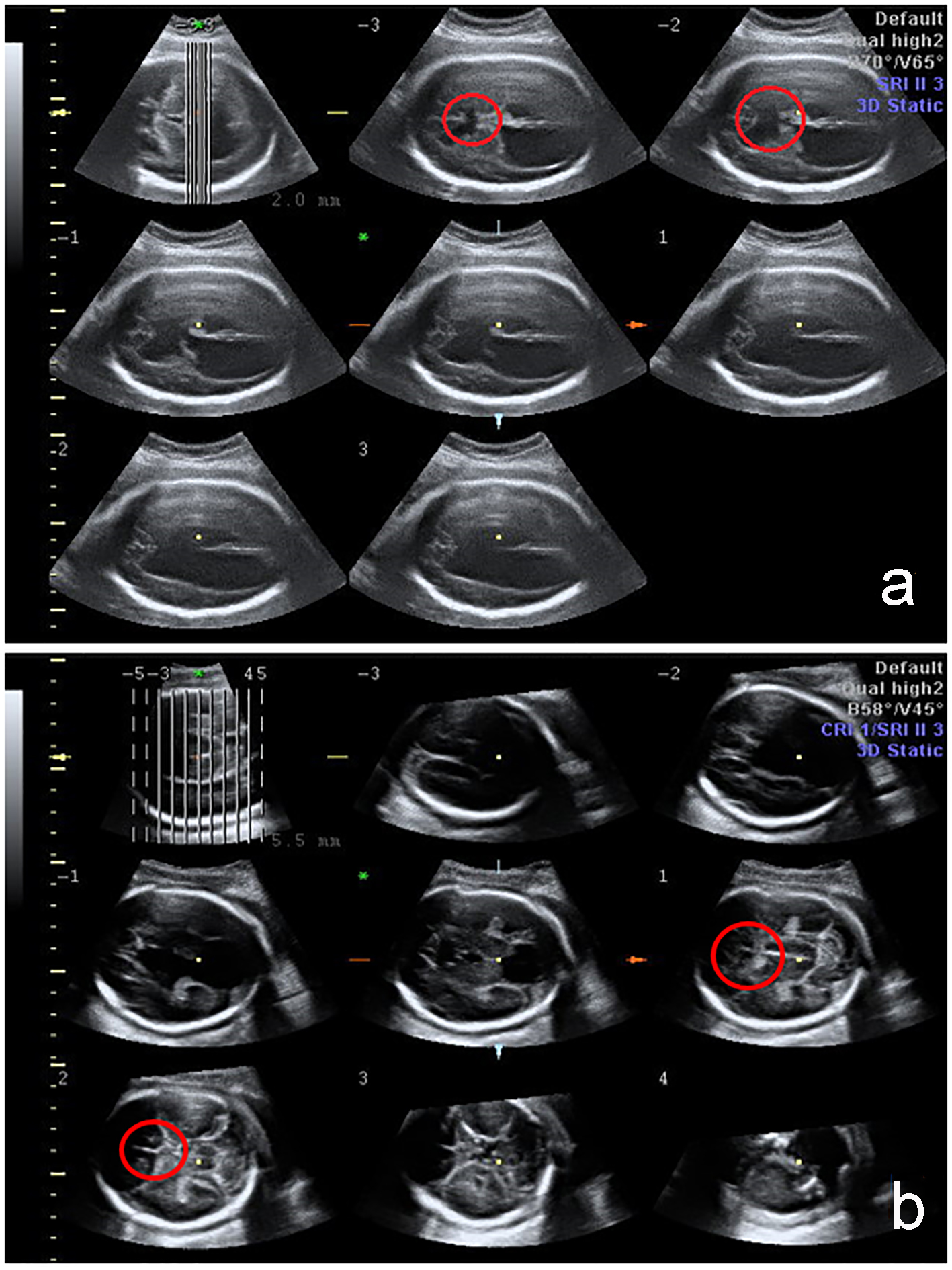 Cliquez pour agrandir l’image |
Figure 1. Évaluation du SNC fœtal en 3D avec l’imagerie échographique tomographique (TUI). L’absence de CSP est indiquée par le cercle rouge dans les cas d’anomalies cérébrales graves : holoprosencéphalie et schizencéphalie. |
Les anomalies du SNC (Fig. 2) associées à l’absence de CSP étaient : agénésie du corps calleux dans sept cas (22,5 %), holoprosencéphalie (HPE) dans trois cas (9.6%), syntélencéphalie dans deux cas (6,4%), porencéphalie dans deux cas (6,4%), hydrocéphalie massive dans cinq cas (16,1%), anencéphalie dans quatre cas (12,9%), schizencéphalie dans deux cas (6,4%), encéphalocèle dans un cas (3,2%) et dysplasie septo-optique dans trois cas (9,6%). La malformation de Dandy-Walker était associée à l’ACC sur trois des sept cas.
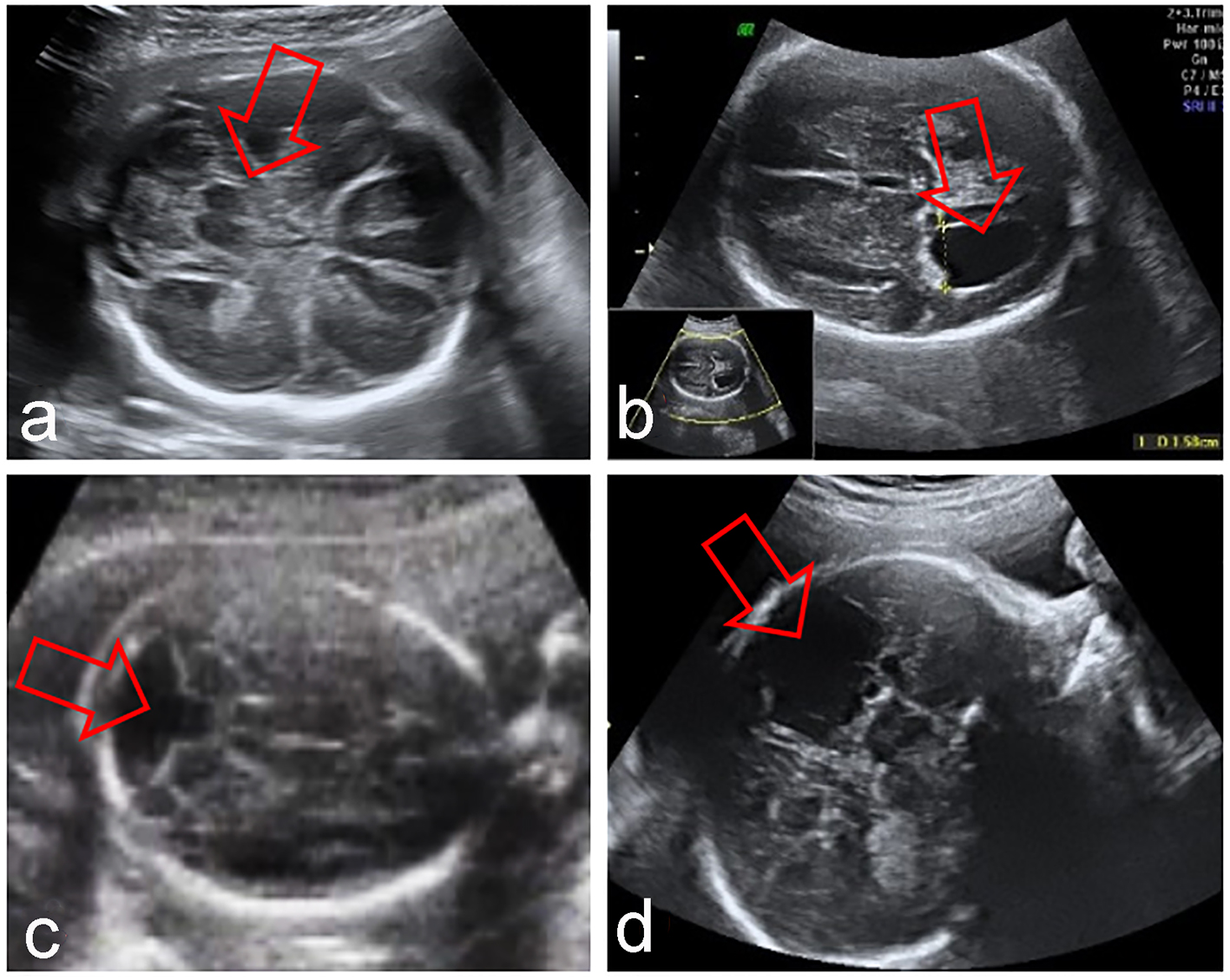 Cliquez pour agrandir l’image |
Figure 2. Anomalies du SNC fœtal associées à une CSP absente : syntélencéphalie (a), hydrocéphalie (b), syndrome de Dandy-Walker (c), porencéphalie (d). |
Dans deux cas, la CSP était absente lors de grossesses après 37 GW (6.4%), lors de leur admission dans notre unité, comme première évaluation imagistique anténatale.
Nous avons considéré un cas de CSP hyperéchogène comme « anormal », car il ne remplit pas les critères normaux de CSP : « une boîte noire entre deux lignes blanches ». Un élargissement persistant de la CSP (Fig. 3), si l’on considère deux déviations standard comme limite de coupure, a été trouvé dans quatre cas. Tous ne présentaient aucune autre anomalie structurelle, et les tests génétiques ont donné des résultats normaux.
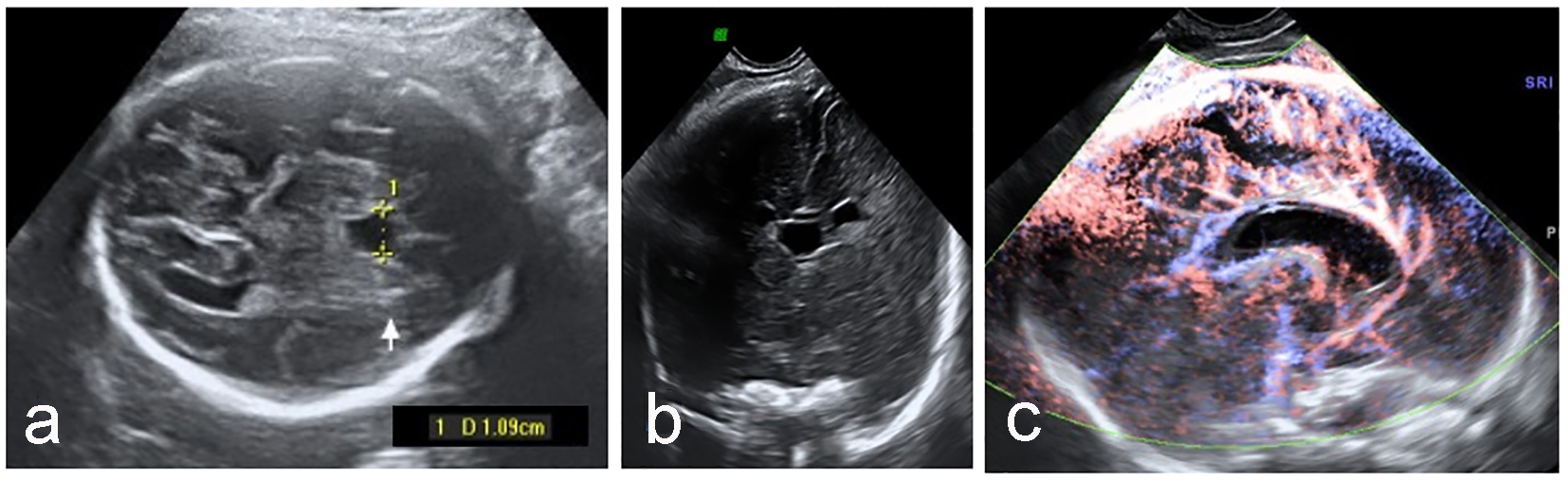 Cliquez pour agrandir l’image |
Figure 3. CSP agrandi (a) avec complexe cérébral antérieur normal dans le plan coronal (b) et artère péricallosale (c). |
Des tests génétiques étaient disponibles dans 24 cas de CSP anormale, et des troubles ont été trouvés dans 16 cas, tous associés à une CSP absente (tableau 1). Genetic disorders were found in 16 cases, half of the cases with absence of CSP. Most frequent genetic disorders were trisomies (21,18,13), but we also found other chromosomal alteration, deletions and translocations.
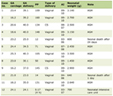 Click to view |
Table 1. Spectrum of CNS Abnormalities and the Associated Abnormalities |
| Discussion | ▴Top |
A normal CSP suggests a normal development of the prozencephalon and may rule out some commonly found CNS structural anomalies. Trois études échographiques, incluant un total de 1024 fœtus normaux, au cours du deuxième et troisième trimestre, ont montré que la CSP peut être visualisée et mesurée chez 100% des fœtus normaux entre 20 et 37 semaines de gestation .
La fermeture de la CSP commence du cavum vaergae postérieur à l’espace réel antérieur de la CSP. Il a été communiqué que le cavum est présent à terme dans 85% des cas et dans 15% à 6 mois après la naissance . Nous avons trouvé deux cas d’absence de CSP dans des examens échographiques réalisés au-delà de 37 semaines. Ces cas ne présentaient pas d’anomalies associées. Leur présentation à l’échographie fœtale était la première pendant la grossesse en cours.
Le développement du CSP est fortement lié au corps calleux, et la plupart des auteurs ont conclu qu’un CSP normal équivaut à un CC normal. Bien que la non-visualisation du CSP ne soit pas synonyme d’agénésie du corps calleux, ces deux entités sont fréquemment associées. Comme le corps calleux n’est pas systématiquement visualisé au cours de l’évaluation de base du SNC lors de l’examen d’anomalie fœtale, l’identification d’une apparence normale de la CSP est d’une importance majeure dans la détection de l’ACC. Pourtant, la CSP peut ne pas être visualisée malgré une CC normale. Comme l’absence de CSP est le plus souvent associée à un ACC, dans de tels cas, la première étape consiste à évaluer le développement de la CC en vue axiale, coronale et sagittale et à visualiser l’artère péricalosale à l’aide d’un examen Doppler dans le plan sagittal. L’IRM et la tractographie sont utiles pour confirmer les aspects échographiques . Dans notre étude, l’absence de CSP était associée au CAC dans sept cas (deux cas d’agénésie partielle et cinq cas d’agénésie totale). Dans quatre de ces cas (57%), des anomalies structurelles non cérébrales ont été décrites et d’autres anomalies du SNC ont été trouvées dans environ 42% de tous les cas. Nous avons trouvé deux cas sur sept avec le syndrome de Dandy-Walker – la plupart des auteurs ont rapporté environ un tiers des cas et dans un cas, un spina bifida était présent.
Une autre anomalie majeure qui bénéficie de la visualisation de la CSP est la dysplasie septo-optique. Dans cette condition, le seul signe anormal de l’échographie prénatale peut être l’absence de CSP, donc l’évaluation de routine de la CSP est très importante pour la détection de ces cas, bien que l’IRM soit cruciale pour la confirmation du diagnostic. Il y avait deux cas de syndrome de Morsier (dysplasie septo-optique) qui associaient l’absence de CSP à une CC normale. La CSP était absente et une légère communication entre les cornes antérieures du ventricule latéral était présente, tandis que la CC était normalement développée. L’état a été confirmé en post-partum et l’un de ces cas a associé une fente labiale unilatérale. Dans l’autre cas, un arc aortique droit avec un canal artériel gauche a été trouvé.
Il y avait 24/36 examens génétiques disponibles (échantillons cytogénétiques et moléculaires). Des troubles ont été trouvés dans 16 cas anormaux de CSP, dont 13 étaient des caryotypes anormaux et les trois autres des résultats anormaux de CGH de réseau moléculaire. L’évaluation génétique est obligatoire surtout en cas d’absence de CSP, le caryotype et le microarray étant également fortement recommandés .
L’absence de CSP peut être associée à un grand nombre de troubles structurels comme l’holoprosencéphalie (HPE), de l’alobare au lobaire, et l’entité de syntélencéphalie décrite plus récemment ; la dysplasie septo-optique (SOD) ; la dysgénésie et l’hypogénèse callosales ; l’hydrocéphalie chronique grave, généralement due à une sténose de l’aqueduc ou à la malformation de Chiari II ; la schizencéphalie ; la porencéphalie, l’hydranencéphalie ; les encéphalocèles basilaires ; et la déficience septale isolée. Dans l’hydranencéphalie, la porencéphalie, la schizencéphalie, l’holoprosencéphalie et l’hydrocéphalie sévère, l’aspect fortement mal structuré du cerveau est évident, et l’anomalie de la CSP n’est qu’une observation, mais avec moins d’importance dans le diagnostic et le pronostic des cas.
La réelle importance clinique d’une CSP prénatale élargie est inconnue, mais il a été souligné que cette constatation doit être suivie d’une recherche détaillée d’anomalies associées et qu’une imagerie postnatale et une évaluation du développement sont indiquées. La persistance d’une CSP large après la naissance a été liée à la schizophrénie si sa largeur dépasse 10 mm, donc un suivi étroit est nécessaire dans ces cas.
La CSP hyperéchogène n’a pas été décrite dans la littérature jusqu’à présent, donc un suivi étroit de notre cas isolé est assez important. Les investigations prénatales étendues de ce cas comme l’IRM et les tests génétiques ont donné des résultats normaux. The infant had a good progress in the first year.
Conclusions
Absent CSP is a very important CNS malformation marker, and its presence should trigger extended fetal morpho-genetic evaluation. In our experience, almost half of the cases associated genetic disorders and 39% associated structural malformations.
Other aspects as isolated enlargement or hyperechoic CSP need further evaluation in larger studies on a long-term outcome in order to establish their significance, as our cases presented a normal postpartum initial evolution, but the number of these abnormalities communicated in the literature is low.
| ▴Top |
- AIUM Practice Guideline for the performance of an antepartum obstetric ultrasound examination. J Ultrasound Med. 2003;22(10):1116-1125.
publié - Ligne directrice de pratique de l’AIUM pour la réalisation d’examens échographiques obstétriques. J Ultrasound Med. 2010;29(1):157-166.
publié - Filly RA, Cardoza JD, Goldstein RB, Barkovich AJ. Détection des anomalies du système nerveux central fœtal : un niveau d’effort pratique pour une échographie de routine. Radiology. 1989;172(2):403-408.
doi pubmed - Nyberg DA. Recommandations pour l’échographie obstétricale dans l’évaluation du crâne fœtal. Radiology. 1989;172(2):309-311.
doi pubmed - Gushiken BJ, Goldstein RB. Approche pratique de l’évaluation de l’axe neural fœtal. Semin Roentgenol. 1999;34(1):5-12.
doi - ACOG Practice Bulletin No. 58. L’échographie pendant la grossesse. Obstet Gynecol. 2004;104(6):1449-1458.
doi - Angtuaco TL. Imagerie ultrasonore des anomalies cérébrales fœtales : trois niveaux anatomiques essentiels. Ultrasound Q. 2005;21(4):287-294.
doi pubmed - Examen sonographique du système nerveux central fœtal : directives pour la réalisation de l' »examen de base » et du « neurosonogramme fœtal ». Ultrasound Obstet Gynecol. 2007;29(1):109-116.
doi pubmed - Callen PW, Callen AL, Glenn OA, Toi A. Columns of the fornix, not to be mistaken for the cavum septi pellucidi on prenatal sonography. J Ultrasound Med. 2008;27(1):25-31.
publié - Timor-Tritsch E. Ultrasonography of the prenatal brain, third edition. McGraw Hill, 2012 ; 2:62-65, 3:128, 6:235-244.
- Sherer DM, Sokolovski M, Dalloul M, Santoso P, Curcio J, Abulafia O. Diagnostic prénatal de la dilatation du cavum septum pellucidum et vergae. Am J Perinatol. 2004;21(5):247-251.
doi pubmed - Bronshtein M, Weiner Z. Prenatal diagnosis of dilated cava septi pellucidi et vergae : associated anomalies, differential diagnosis, and pregnancy outcome. Obstet Gynecol. 1992;80(5):838-842.
publié - Jou HJ, Shyu MK, Wu SC, Chen SM, Su CH, Hsieh FJ. Mesure échographique du cavum septi pellucidi du fœtus. Ultrasound Obstet Gynecol. 1998;12(6):419-421.
doi pubmed - Winter TC, Kennedy AM, Byrne J, Woodward PJ. Le cavum septi pellucidi : pourquoi est-il important ? J Ultrasound Med. 2010;29(3):427-444.
publié - Falco P, Gabrielli S, Visentin A, Perolo A, Pilu G, Bovicelli L. Echographie transabdominale du cavum septum pellucidum chez les fœtus normaux aux deuxième et troisième trimestres de la grossesse. Ultrasound Obstet Gynecol. 2000;16(6):549-553.
doi pubmed - Serhatlioglu S, Kocakoc E, Kiris A, Sapmaz E, Boztosun Y, Bozgeyik Z. Mesure échographique du cervelet fœtal, du cisterna magna et du cavum septum pellucidum chez les fœtus normaux aux deuxième et troisième trimestres de la grossesse. J Clin Ultrasound. 2003;31(4):194-200.
doi pubmed - Paladini D, Pastore G, Cavallaro A, Massaro M, Nappi C. Agénésie du corps calleux fœtal : les signes échographiques changent avec l’avancement de l’âge gestationnel. Ultrasound Obstet Gynecol. 2013;42(6):687-690.
doi pubmed - Mitter C, Prayer D, Brugger PC, Weber M, Kasprian G. Tractographie in vivo des fibres d’association fœtales. PLoS One. 2015;10(3):e0119536.
doi pubmed - Sarwar M. Le septum pellucidum : normal et anormal. AJNR Am J Neuroradiol. 1989;10(5):989-1005.
publié
Il s’agit d’un article en accès libre distribué selon les termes de la licence internationale Creative Commons Attribution-NonCommercial 4.0, qui permet l’utilisation, la distribution et la reproduction non commerciale sans restriction sur n’importe quel support, à condition que l’œuvre originale soit correctement citée.
Journal of Clinical Gynecology and Obstetrics est publié par Elmer Press Inc.