INTRODUCTION
Les pneumopathies interstitielles forment un groupe extrêmement hétérogène de troubles qui ont en commun l’existence d’une composante inflammatoire du parenchyme pulmonaire pouvant éventuellement évoluer vers une fibrose et provoquant, d’un point de vue fonctionnel, une altération restrictive pouvant éventuellement conduire à une insuffisance respiratoire.
Bien qu’il s’agisse de maladies dont le diagnostic et la prise en charge sont effectués dans le domaine spécialisé de la pneumologie, le traitement est fondamentalement suivi en ambulatoire, et étant donné que celui-ci peut et est souvent ponctué de complications diverses, il est important que le médecin de premier recours connaisse les directives actuelles pour le traitement de ces maladies, car à de nombreuses occasions, il sera consulté par des patients présentant ce type de problème, que ce soit en raison de complications respiratoires ou d’autres types de complications.
Le but de cette revue est de mettre à jour les modalités thérapeutiques qui peuvent être proposées aujourd’hui à ces patients. Nous pensons qu’il est de la plus haute importance non seulement que le médecin de premier recours ait une connaissance actualisée de ce sujet, mais aussi qu’il collabore avec le pneumologue afin d’obtenir une bonne observance du traitement du patient et une détection et un traitement précoces des complications qui peuvent survenir.
Parmi les maladies interstitielles, la plus pertinente en raison de sa fréquence et de sa gravité est la fibrose pulmonaire idiopathique (FPI). En revanche, son traitement est le modèle sur lequel repose le traitement de la quasi-totalité de ces maladies, à l’exception de celles pour lesquelles on peut trouver un agent étiologique connu, puisque dans ces dernières, l’élimination de cet agent sera la base fondamentale de la thérapie. Nous nous concentrerons donc sur un examen des directives actuelles pour la gestion de la FPI. Nous allons d’abord rappeler brièvement ses caractéristiques cliniques, puis développer en détail la prise en charge actuelle de la FPI.
DESCRIPTION CLINIQUE
La FPI représente probablement le prototype le plus agressif des pneumopathies interstitielles diffuses. Sa prévalence a été estimée par certains auteurs à 5 cas pour 100 000 habitants1, bien qu’elle soit en réalité inconnue. Sa pathogenèse est liée à des altérations des voies complexes du système immunitaire2,3. 2,3 Pour cette raison, la plupart des médicaments utilisés sont des agents immunosuppresseurs.
La présentation clinique la plus courante est une dyspnée d’apparition insidieuse accompagnée d’une toux sèche, qui peut être le symptôme le plus gênant pour le patient en raison de sa nature paroxystique et essoufflée. En général, la dyspnée est initialement sans particularité et un indice de suspicion élevé est nécessaire pour un diagnostic précoce. Parfois, l’évolution est rapide dès le début et conduit en quelques mois à une insuffisance respiratoire irréversible, comme c’était le cas dans les descriptions initiales de Hamman et Rich. Cependant, le plus souvent, l’affection est présente depuis des mois au moment où le patient effectue sa première visite.
À l’examen physique, des crépitants fins et secs (râles de velcro) sont très caractéristiques et peuvent être entendus dans plus de 80 % des cas. Dans les cas avancés, on peut observer une dyspnée à l’effort léger, voire une cyanose labiale. Des acropaquies peuvent être observées dans environ 30 à 50 % des cas. La suspicion de FPI est renforcée lorsque des signes tels qu’un motif réticulaire, des zones d’infiltration en verre dépoli et une diminution du volume pulmonaire apparaissent à la radiographie pulmonaire et au scanner. Enfin, l’examen fonctionnel confirmera l’existence d’un schéma restrictif et l’analyse des gaz du sang artériel confirmera l’existence d’une hypoxémie. Dans les cas douteux, la démonstration d’une hypoxémie à l’effort est d’une grande valeur.
La confirmation définitive du diagnostic ne peut se faire que par une biopsie pulmonaire. Cependant, la réalité est que même dans de nombreuses séries publiées, les cas sans diagnostic histologique prédominent. Récemment, l’American Thoracic Society (ATS) a proposé que l’existence de tous ces critères signifie que, en l’absence d’exposition à des médicaments, à des éléments environnementaux ou à une maladie du tissu conjonctif, un diagnostic de FPI peut très probablement être posé en l’absence de biopsie pulmonaire4 dans les cas où la biopsie pulmonaire représenterait un risque inacceptable.
La pneumonie interstitielle habituelle (PIE) est le schéma histopathologique qui permet d’identifier ces patients4. Ceux présentant des tableaux de pneumonie interstitielle desquamative, de maladie interstitielle associée à une bronchiolite respiratoire, de pneumonie interstitielle non spécifique, de pneumonie interstitielle lymphoïde, de pneumonie interstitielle aiguë et de bronchiolite oblitérante avec pneumonie organisatrice sont considérés comme des entités différentes.
Une fois le diagnostic et la gravité clinique établis, la plupart des patients ont besoin d’un traitement car il s’agit d’une maladie mortelle sans rémission spontanée. La réponse au traitement est faible. La survie à cinq ans est de 50 %5,2,6-8. Le traitement nécessite environ 3 à 6 mois avant que son efficacité ne soit établie. Les rapports sur le traitement de ces maladies sont rares : il s’agit d’essais thérapeutiques non contrôlés portant le plus souvent sur un petit nombre de patients9.
Malgré l’inévitable progression insidieuse et irréversible de la maladie, le nihilisme thérapeutique n’est pas justifié. Il existe des exemples de fibrose non traitée dont la progression lente conduit à une fibrose irréversible ne répondant pas au traitement ou développant une phase rapidement progressive. Toutes nécessitent probablement un traitement au moment de la présentation. Toutefois, les patients présentant des contre-indications au traitement ou ceux qui ne présentent aucun signe de déficience fonctionnelle peuvent suivre un programme de suivi à intervalles de 3 à 6 mois. La prise en charge des patients âgés présentant une histologie à prédominance de fibrose et une grave altération de la fonction pulmonaire est également difficile. Ces patients reçoivent un traitement court de 3 à 6 mois pour déterminer la réversibilité. Peu d’entre eux auront une réponse positive.
Il est souvent difficile de déterminer si le traitement produit l’effet désiré. Une amélioration subjective se produit souvent (jusqu’à 70 % des patients). Une amélioration objective se produit dans 20 à 30 % des cas. Les paramètres les plus couramment utilisés pour définir une amélioration fonctionnelle (objective) sont une augmentation de la capacité pulmonaire totale (CPT) ou de la capacité vitale (CV), une augmentation de la capacité de diffusion et une réduction ou une normalisation du déclin de la SaO2 pendant l’exercice4,10-12.
Traitement
Corticostéroïdes
Les corticostéroïdes sont le pilier du traitement de la FPI, bien que leur succès soit plutôt faible : une réponse favorable survient chez un tiers des patients. La raison de leur utilisation est de supprimer l’alvéolite chronique. On pense qu’ils agissent en supprimant la migration des neutrophiles et des lymphocytes dans le poumon, en diminuant la concentration des immunocomplexes, en modifiant la fonction des macrophages alvéolaires et l’adhésion des neutrophiles aux surfaces endothéliales.
On ne sait pas pourquoi certains patients répondent et d’autres pas. Elle pourrait être liée à la présence de récepteurs de surface sur les cellules du parenchyme pulmonaire. Une courte durée des symptômes, une atteinte fonctionnelle et radiologique légère, le sexe féminin et l’âge plus jeune des patients semblent être associés à un meilleur résultat.
Toutefois, bien qu’il s’agisse du traitement le plus largement utilisé, sa performance à long terme n’a été confirmée par aucune étude prospective, en simple aveugle et contrôlée par placebo.
Izumi et al ont analysé la survie de 222 patients et ont constaté qu’à 10 ans, il n’y avait pas de différence de survie entre ceux qui avaient reçu des stéroïdes et les patients non traités13. Néanmoins, lorsqu’un patient atteint de FPI présente une fonction pulmonaire altérée ou des symptômes qui interfèrent avec la vie normale, il est raisonnable de tenter une corticothérapie. En effet, l’amélioration spontanée de la maladie est très rare, et s’il est décidé de retarder l’initiation du traitement, cela doit se faire au prix d’une surveillance étroite du patient afin de pouvoir initier le traitement dès que la détérioration est évidente.
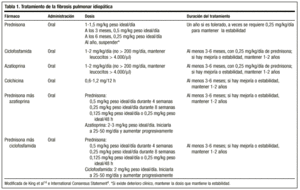
Le traitement classique commence par la prednisone 1,5-2 mg/kg de poids idéal/jour en une seule prise le matin, sans dépasser une dose de 100 mg. Après 2 à 3 mois, le patient est réévalué par une radiographie et une étude fonctionnelle. Si le patient répond, la prednisone est diminuée (1 à 2 mg/semaine) jusqu’à une dose d’entretien de 0,5 mg/kg de poids idéal/jour, qui peut être réduite par la suite. En l’absence de réponse, un médicament immunosuppresseur est ajouté. En général, il est peu probable que le traitement puisse être arrêté, et des doses de 0,25 mg/kg de poids idéal/jour sont habituellement maintenues.
Récemment, les sociétés thoraciques américaine et européenne, afin d’éviter les effets secondaires de ce médicament et d’obtenir de meilleurs taux de survie, sont parvenues à un consensus pour commencer le traitement par une combinaison de prednisone ou d’un autre corticostéroïde à des doses équivalentes et d’un immunosuppresseur (azathioprine ou cyclophosphamide)4 : prednisone 0,5 mg/kg de poids idéal/jour pendant 4 semaines ; 0,25 mg/kg de poids idéal/jour pendant 8 semaines, puis diminution progressive jusqu’à 0,125 mg/kg de poids idéal/jour ou 0,25 mg/kg de poids idéal/48 heures.
Il n’existe pas de directives sur la durée du traitement ; la durée minimale est d’un an. Il est possible qu’après des mois ou des années, la maladie se réactive, auquel cas un traitement avec les mêmes agents doit être initié. Dans tous les cas, la réponse au traitement doit toujours être évaluée en termes d’amélioration objective, et la possibilité de maintenir le patient de façon chronique à une dose garantissant la stabilité doit être réservée aux cas où il y a une amélioration avec les corticostéroïdes, une détérioration lors de la diminution progressive et une nouvelle amélioration lors d’une nouvelle augmentation de la dose. La dose requise pour chaque patient ne peut être déterminée qu’au cas par cas, en fonction de la réponse aux modifications de la dose. De nombreux auteurs considèrent qu’un traitement à vie est nécessaire.14
Lorsque la maladie est initialement sévère, des corticostéroïdes à forte dose ont été proposés. Keogh et al ont utilisé des corticostéroïdes parentéraux intermittents à forte dose chez des patients atteints d’une maladie grave : méthylprednisolone intraveineuse, 2 g une fois par semaine, plus prednisone orale, 0,25 mg/kg/jour. La raison en est qu’ils ont un effet plus prononcé sur l’alvéolite neutrophilique (associée à un mauvais pronostic) que les stéroïdes à faible dose15. Cependant, ce travail souffre de certaines lacunes, comme le suivi court de 6 mois, le petit nombre de patients et les doses de prednisone inférieures à la norme9. Le traitement initial par corticostéroïdes intraveineux à forte dose (méthylprednisolone 250 mg/6 h) a été utilisé dans le but de contrôler la maladie le plus tôt possible lorsqu’elle est rapidement progressive. Il n’y a pas d’études contrôlées pour déterminer si ces méthodes sont efficaces ; cependant, des doses plus élevées semblent avoir une plus grande efficacité, bien que les effets secondaires soient également plus importants.14.
Les AGENTS CITOTOXIQUES
ont été classiquement utilisés comme une alternative aux corticostéroïdes lorsque ceux-ci étaient inefficaces, et aussi lorsque leurs effets secondaires empêchent leur administration ultérieure. Conformément à l’esprit éminemment pratique de cet article, nous passerons en revue les agents qui se sont avérés utiles en pratique clinique, même si, comme dans le cas des corticostéroïdes, nous manquons de longues séries prospectives et randomisées qui nous permettent de savoir exactement quel est leur rôle exact dans le traitement de la FPI.
Azathioprine
Son mécanisme d’action repose sur la substitution des purines dans la synthèse de l’acide désoxyribonucléique, inhibant ainsi l’adénine désaminase et inactivant les lymphocytes. Les autres effets comprennent la suppression de l’activité des cellules tueuses naturelles, de la production d’anticorps et de la cytotoxicité cellulaire dépendante des anticorps.
La dose recommandée est de 2 mg/kg/jour, sans dépasser 200 mg/jour, pendant une période de 3 à 6 mois. L’administration est initiée à la dose de 50 mg/jour et, si elle est bien tolérée, la dose est augmentée à intervalles de 1 à 2 semaines jusqu’à ce que la dose prévue soit atteinte.
Les principaux effets secondaires sont hématologiques : leucopénie, anémie, thrombocytopénie, érythroblastopénie et pancytopénie. Il est nécessaire de surveiller la numération leucocytaire, et si elle tombe en dessous de 4 000/μl, l’azathioprine doit être interrompue jusqu’à ce que ce chiffre remonte. En outre, des troubles gastro-intestinaux tels que nausées et vomissements, ulcère gastroduodénal, diarrhée et légère élévation des enzymes hépatiques peuvent survenir. Globalement, dans jusqu’à 20 à 30 % des cas, les effets secondaires nécessitent l’arrêt de l’azathioprine.
Raghu et al ont comparé l’efficacité des glucocorticoïdes à forte dose par rapport à l’azathioprine en association avec des glucocorticoïdes à faible dose. L’utilisation de ce médicament s’est accompagnée d’une modeste amélioration des paramètres de la fonction respiratoire et d’une augmentation de la survie17.
Cyclophosphamide
Est un agent alkylant. Son mode d’action est la déplétion lymphocytaire et supprime donc la fonction lymphocytaire. La dose recommandée est de 2 mg/kg/jour en une seule prise orale, généralement envisagée avec 0,25 mg/kg/jour de prednisone orale, sans dépasser 200 mg/jour pendant au moins 3 mois et jusqu’à 9-12 mois.
L’effet secondaire le plus important est la leucopénie. Un taux de leucocytes totaux supérieur à 3 000/μl ou un taux de neutrophiles supérieur à 1 500/μl doit être maintenu. Les autres complications comprennent la thrombocytopénie, l’hématurie secondaire à la cystite hémorragique, l’anorexie, les nausées et les vomissements, la suppression de la moelle osseuse, l’azoospermie et l’aménorrhée, l’infection et la malignité hématologique.
Kolb et al ont étudié 18 patients atteints de FPI pour déterminer l’efficacité et la sécurité d’un traitement pulsé au cyclophosphamide. La cyclophosphamide a été administrée par intermittence (1-1,3 g/mois) avec de la prednisolone par voie orale pendant un an. Après cette période, le cyclophosphamide a été interrompu et les doses atteintes de corticostéroïdes ont été poursuivies8 . Ils ont été suivis pendant un minimum de 14 mois, en contrôlant la qualité de vie, le TLC, le VEMS, la capacité de transfert du CO (DLCO), la SaO2, ainsi que les symptômes. Un patient a dû être interrompu en raison de pneumonies récurrentes ; 11 patients ont été considérés comme répondeurs (5 améliorés et 6 stabilisés) et 6 se sont détériorés. La toxicité à long terme du cyclophosphamide dépend des doses cumulées. Les doses intermittentes sont aussi efficaces et mieux tolérées que les doses quotidiennes. Les répondeurs avaient un pourcentage plus élevé de lymphocytes dans le LBA. Un autre résultat notable est que la QDV initiale des répondeurs était significativement meilleure en raison d’un temps plus court avant la progression, ce qui suggère que ce traitement devrait être commencé tôt.
Baughman et Lower ont traité 33 patients atteints de FPI avec 1 000-1 800 mg de cyclophosphamide/2 semaines pendant 18 mois. Ceux qui ont survécu 18 mois présentaient une amélioration significative de la FVC qui s’est maintenue au cours de l’année suivante. Une réduction de la prednisolone a été possible de 32 à 4 mg/jour16.
Pénicillamine
Agent antifibrosant. Il a été suggéré qu’en interférant avec la formation de liaisons covalentes intra- et intermoléculaires des fibres de collagène, il inhibe le développement de la fibrose pulmonaire. Certains cas de maladies du tissu conjonctif avec fibrose pulmonaire associée ont été décrits dans lesquels il a eu une certaine efficacité, mais il n’existe pas d’études contrôlées sur de grandes séries de patients, de sorte qu’il ne peut actuellement être considéré comme un traitement alternatif valable. Des études complémentaires sont donc nécessaires avant de pouvoir formuler une recommandation.
Colchicine
Diminue la sécrétion de collagène et augmente l’activité collagénolytique. Il a également un effet anti-inflammatoire. Des études sont nécessaires pour déterminer plus précisément son rôle dans le traitement de cette maladie, mais il existe des études dans lesquelles on lui a attribué une efficacité similaire à celle des corticostéroïdes, et ses effets secondaires ne sont pas graves. Il est recommandé à la dose de 0,6 mg, une ou deux fois par jour, chez les patients réfractaires aux corticostéroïdes, seuls ou en association avec des agents immunosuppresseurs. Bien que certains auteurs la placent en première ligne de traitement, nous manquons actuellement de données pour l’établir comme telle (tableau 1).
Transplantation pulmonaire
Les cas où la maladie évolue sans réponse au traitement et finit par provoquer une insuffisance respiratoire sont malheureusement les plus nombreux dans cette maladie. Il n’y a actuellement aucune perspective de traitement médical pour ces patients, si ce n’est la palliation. Dans ce contexte sombre, la possibilité de transplantation pulmonaire a émergé dans les années 1980 et s’est développée au point de devenir une option thérapeutique qui doit être considérée comme un outil à usage clinique et qui n’est plus limitée au domaine expérimental. Son indication la plus appropriée est chez les personnes de moins de 65 ans, en l’absence de maladie progressive et irréversible dans d’autres organes, ainsi que d’infection active non résolue ou d’infection par des germes multirésistants. Une prédisposition personnelle et un état nutritionnel adéquats sont également nécessaires. Les patients dont le CPT est inférieur à 60 % de la valeur attendue ou dont la capacité de diffusion pulmonaire est inférieure à 40 % ont une survie inférieure à 2 ans. A ce degré de dysfonctionnement, ils nécessitent une oxygénothérapie et se désaturent facilement avec un exercice minimal. La transplantation pulmonaire doit être envisagée dans ces cas, dès que l’oxygénothérapie devient nécessaire et que l’on vérifie la progression inéluctable de la maladie. Le type de transplantation de choix est la transplantation d’un poumon, réservant la transplantation de deux poumons aux cas où il y a des doutes sur le comportement du poumon résiduel. Lorsque le patient est âgé de moins de 50 ans et qu’il présente une insuffisance cardiaque droite, une transplantation cardio-pulmonaire peut être envisagée. Cependant, dans cette situation, la maladie est très avancée et il est peu probable qu’elle résiste à l’attente18,19. La survie post-transplantation devrait être de 80 % la première année et de 50 à 60 % à 5 ans, ce qui améliore considérablement le pronostic d’un patient atteint de FPI ayant atteint l’insuffisance respiratoire.
ÉVALUATION DE LA RÉPONSE AU TRAITEMENT
En l’absence de complications, il peut s’écouler jusqu’à 6 mois avant d’obtenir une quelconque réponse au traitement. Après cette période, si le patient s’est amélioré ou est stable, les mêmes doses doivent être maintenues. S’ils se sont aggravés, il faut passer à un autre traitement (un autre agent immunosuppresseur ou même une transplantation). A 12 mois, il doit être réévalué de la même manière et après 18 mois, individualisé en fonction de la réponse. Se recomienda continuar indefinidamente sólo en aquellos pacientes con mejoría continua o estabilización (tabla 2).

Una respuesta favorable se define por dos o más de los siguientes criterios en dos visitas consecutivas:
1. Mejoría de los síntomas, sobre todo en el grado de esfuerzo requerido para que el paciente deba parar cuando hace un esfuerzo o un descenso en la frecuencia o gravedad de la tos.
2. Reducción de las alteraciones radiológicas.
3. Mejoría funcional definida por dos o más de los siguientes parámetros:
Aumento >= 10% en la TLC o CV (o >= 200 ml).
Aumento >= 15% en DLCO o >= 3 ml/min/mmHg.
Mejoría o normalización de la SaO2 o pO2 (aumento >= 4% o >= 4 mmHg) durante un prueba de esfuerzo cardiorrespiratoria.
Una respuesta estable (también considerada favorable) se define por dos o más de los siguientes criterios en dos visitas consecutivas:
1. Cambio de
2. Cambio de
3. Sin cambios en la SaO2 o pO2 (aumento
Un fracaso del tratamiento se define por dos o más de los siguientes criterios en dos visitas consecutivas:
1. Aumento de los síntomas, sobre todo disnea o tos.
2. Aumento de las alteraciones radiológicas, especialmente imágenes en panal o signos de hipertensión pulmonar.
3. Deterioro funcional definido por dos o más de los siguientes parámetros:
Descenso >= 10% en la TLC o CV (o >= 200 ml).
Descenso >= 15% en DLCO o >= 3 ml/min/mmHg.
Augmentation de la SaO2 (= 4%) ou élévation du gradient alvéolo-artériel (AapO2) (= 4 mmHg) au repos ou lors d’une épreuve d’effort cardiorespiratoire.
CONCLUSIONS
La fibrose pulmonaire idiopathique est une maladie mortelle, sans rémission spontanée et répondant mal au traitement. Les études sur ce dernier point sont rares et portent sur un petit nombre de patients. Les corticostéroïdes maintenus pendant au moins un an à différentes doses constituent le régime le plus accepté, seuls ou en association avec d’autres agents immunosuppresseurs. Récemment, l’American Thoracic Societies et l’European Respiratory Societies ont préconisé une thérapie combinée4 . Des agents anti-fibrosants sont à l’étude dans le but d’empêcher le processus fibrotique de s’installer et donc d’être irréversible, car de nombreux patients sont diagnostiqués à des stades tardifs. Compte tenu des taux de survie et de l’évolution, la transplantation pulmonaire doit être envisagée dès que les patients échouent au traitement et nécessitent une oxygénothérapie. Tout ceci suggère la nécessité d’établir des études multicentriques dans l’idée de recruter un nombre suffisant de patients pour évaluer l’efficacité des traitements administrés, ainsi que pour identifier l’histoire naturelle de la maladie et l’incidence et la prévalence réelles4,20. D’autre part, il est recommandé d’identifier et d’initier un traitement dans les premiers stades de la maladie afin d’obtenir des taux de survie plus élevés.