- Introduction
- Anyagok és módszerek
- Mintaelőkészítés
- MinION könyvtárkészítés és szekvenálás
- Genomikai elemzés
- Eredmények
- MinION szekvenálási adatok és a vírusgenomok összeállítása
- A keresztmintás szennyeződés mértéke és forrása
- Megbeszélés
- A szerzők hozzájárulása
- Finanszírozás
- Érdekütközéssel kapcsolatos nyilatkozat
- Köszönet
- Kiegészítő anyagok
- Lábjegyzet
Introduction
A metagenomikai szekvenálás lehetővé teszi a kórokozók elfogulatlan azonosítását klinikai mintából. Azt ígéri, hogy egyetlen és univerzális próbaként szolgálhat a fertőző betegségek diagnosztikájához közvetlenül a mintákból, a priori ismeretek nélkül (Bibby, 2013; Miller és mtsai., 2013; Schlaberg és mtsai., 2017). A kórokozó fajok azonosítása mellett a széles és mély metagenomikai szekvenciaadatok a kezelés és a prognózis meghatározása, a járványkitörések felderítése és a fertőzés epidemiológiájának nyomon követése szempontjából releváns információkat szolgáltathatnak (Greninger et al., 2010; Yang et al., 2011; Qin et al., 2012; Loman et al., 2013). Az újgenerációs szekvenáló (NGS) platformok szerény költségek mellett hatalmas adatátviteli teljesítményt képesek produkálni, azonban a klinikai diagnosztikában és a közegészségügyben való alkalmazását eddig a bonyolultság, a lassúság és a tőkebefektetés korlátozta.
A MinION egy tenyérnyi méretű, valós idejű, egymolekulás genomszekvenáló, amelyet az Oxford Nanopore Technologies (ONT) fejlesztett ki. A MinION kompakt mérete és valós idejű jellege megkönnyítheti a metagenomikai szekvenálás alkalmazását a fertőző betegségek point-of-care vizsgálatában, amint azt több proof-of-concept vizsgálat is bizonyította, beleértve a Chikungunya (CHIKV), az Ebola (EBOV) és a hepatitis C vírus (HCV) azonosítását emberi klinikai vérmintákból, célpont dúsítás nélkül (Greninger et al., 2015), valamint bakteriális kórokozók kimutatása vizeletmintákból (Schmidt et al., 2016) és légúti mintákból, előzetes tenyésztés nélkül (Pendleton et al., 2017).
A MinION adatátviteli teljesítménye jelentősen megnőtt a 2015-ös megjelenése óta, és ma már minden egyes fogyasztható áramlási cella akár 10-20 Gb DNS-szekvenciaadatot is generál. Ez lehetővé teszi a felhasználók számára az áramlási cella hatékonyabb kihasználását (és a költségek csökkentését) azáltal, hogy egyetlen szekvenálási futtatás során több mintát multiplexelnek. Az ONT olyan PCR-mentes vonalkódkészleteket fejlesztett ki, amelyek akár 12 minta multiplexelését is lehetővé teszik.
A több légúti mintában lévő influenza A vírus kimutatása lehet a multiplexelt MinION szekvenálási próba egyik diagnosztikai felhasználási módja. A potenciálisan széles vírustiter-tartományt tartalmazó minták közvetlen szekvenálásakor azonban fontos tisztában lenni a minták keresztkontaminációjának lehetőségével, mind a könyvtárkészítés, mind a szekvenálást követő bioinformatikai vonalkód-demultiplexelési szakasz során. A következőkben bemutatunk egy egyedülálló MinION szekvenálási adathalmazt és a multiplex szekvenálás során a kereszt-vonalkódszennyeződés mértékének és forrásának vizsgálatának eredményeit.
Anyagok és módszerek
Mintaként egy influenza A vírussal fertőzött görény orrmosási mintát használtunk, valamint két aliquot negatív, nem fertőzött görényből származó orrmosási mintát (egy nem kapcsolódó vizsgálatból származó, már meglévő, nem használt állomány) külön dengue és chikungunya vírussal spicceltünk. E vírusok egyike sem releváns a klinikai diagnosztika szempontjából a légúti mintákban, de itt egyértelmű, különálló markerként szolgálnak a keresztmintás szennyeződés értékeléséhez. A szekvenáló könyvtárakat minden egyes mintához párhuzamosan készítettük el, egy negatív orrmosási kontrollal együtt, vonalkódoltuk és külön-külön szekvenáltuk. Ezután a szekvenáló könyvtárak egy aliquotját összevontuk, és multiplex MinION-szekvenálást végeztünk. A négy egyéni futtatásból (a továbbiakban “CHIKV”, “DENV”, “FLU-A” és “Negatív”) és a multiplex futtatásból (a továbbiakban “Multiplexelt”) származó leolvasásokat ezután elemeztük, hogy megvizsgáljuk a keresztminta-kontamináció mértékét és forrását.
Mintaelőkészítés
A projekt engedélyét a helyi AWERB (Animal Welfare and Ethics Review Board) felülvizsgálta, majd a Home Office engedélyezte. Az RNS-t a QIAamp viral RNA kit (Qiagen) segítségével, a gyártó utasításainak megfelelően, az influenza A (H1N1) vírust (A/California/04/2009) tartalmazó görény orrmosásból és egy negatív orrmosási mintacsoportból vontuk ki. A negatív minták kivonatának aliquotjait a The National collection of Pathogenic Viruses1 vírusgyűjteményből származó dengue (DENV) (TC861HA törzs, GenBank: MF576311) vagy CHIKV (S27 törzs, GenBank: MF580946.1) vírus-RNS-szel spicceltük. A mintákat DNázzal kezeltük TURBO DNase (Thermo Fisher Scientific, Waltham, MA, Egyesült Államok) segítségével, és az RNA Clean & ConcentratorTM-5 kit (Zymo Research) segítségével tisztítottuk. cDNS-t készítettünk és a korábban leírtak szerint módosított Sequence-Independent-Single-Primer-Amplification módszerrel (Greninger et al., 2015) sokszorosítottuk (Atkinson et al., 2016). Az amplifikált cDNS-t a Qubit dsDNS HS Assay Kit (Thermo Fisher Scientific, Waltham, MA, Egyesült Államok) segítségével számszerűsítettük, és 1 μg-ot használtunk inputként minden egyes MinION könyvtárkészítéshez, kivéve a negatív kontrollt, ahol a teljes mintát (32 ng) használtuk.
MinION könyvtárkészítés és szekvenálás
A Ligation Sequencing Kit 1D (SQK-LSK108) és a Native Barcoding Kit 1D (EXP-NBD103) az ONT standard protokolljainak megfelelően került felhasználásra, azzal a különbséggel, hogy mind a négy könyvtárkészítménybe csak egy-egy vonalkód került. Mindegyik könyvtárat külön áramlási cellán futtattuk, és a négy külön-külön vonalkódolt könyvtár egyesítésével egy ötödik összevont könyvtárat készítettünk. A könyvtárakat R9.4 áramlási cellákon szekvenáltuk. A vizsgálat felépítése az 1. ábrán látható.
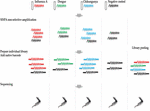
1. ÁBRA. A vizsgálati terv áttekintése. RNS-t négy mintából vontunk ki, köztük egy influenza A vírussal fertőzött görény orrmosási mintából, két negatív, dengue- és chikungunya-vírussal spiccelt görény orrmosási mintából, valamint egy negatív görény orrmosási kontrollból. cDNS-t készítettünk és amplifikáltunk szekvenciafüggetlen-egyszeres-primer-amplifikációs módszerrel. Az egyes minták szekvenáló könyvtárait párhuzamosan készítettük el, vonalkódoltuk, és egyedi áramlási cellákon szekvenáltuk. A multiplex szekvenálást a négy egyedi könyvtár összevonásával is elvégeztük. A négy egyedi futtatásból és a multiplex futtatásból származó leolvasásokat elemeztük, hogy felmérjük a multiplex szekvenálás során a kereszt-vonalkódszennyezés mértékét és forrását.
Genomikai elemzés
A leolvasásokat az Albacore v2.1.7 (ONT) segítségével, vonalkód-demultiplexálással báziskódoltuk. Az egyes szekvenálási futtatásokból származó leolvasásokat a Minimap2 (Li, 2018) segítségével képeztük le az egyes vírusok genomi szekvenciáira. A referenciához leképezett olvasatok számát a Pysam2 segítségével számoltuk meg. A de novo assembly-t a Canu v1.7 (Koren et al., 2017) segítségével végeztük el, és az így kapott genomtervezetet a Nanopolish (Mongan et al., 2015) segítségével csiszoltuk a jelszintű adatokkal.
A multiplex MinION szekvenálási adatok szigorú vonalkód-demultiplexálásának lehetővé tétele érdekében két körös elemzést végeztünk a Porechop (v0.2.23) segítségével. Az adapterszekvencia jelenléte a leolvasás közepén a kiméra jele. A Porechop segítségével minden egyes olvasatot megvizsgáltunk, és azokat, amelyek középső régiója >75%-os azonosságot mutat az adapterszekvenciával, kimérikus olvasatként azonosítottuk. A Porechopban beállítottuk a “-middle_threshold” opciót, és 75-ös küszöbértéket választottunk. A második körben a Porechop segítségével kerestük a vonalkódszekvenciát a leolvasás elején és végén is; a leolvasásokat csak akkor soroltuk be, ha a két végén ugyanazt a vonalkódot találtuk. A Porechopban beállítottuk a “-require_two_barcodes” opciót, és a vonalkód-pontszám küszöbértékét 70-ben határoztuk meg. A kimérikus olvasatok potenciális szignatúrájának megtalálása érdekében megvizsgáltuk a MinION szekvenáló által a FAST5 fájlban tárolt olvasási áramjeleket. Az áramjeleket az ONT fast5 API4 segítségével extraháltuk, és az R5-ben implementált ggplot2 segítségével ábrázoltuk a kimérikus és nem kimérikus olvasatok összehasonlításához.
Eredmények
MinION szekvenálási adatok és a vírusgenomok összeállítása
A futási idő különbségei miatt az egyes MinION szekvenálási futások teljesítménye eltérő volt. A maximális ∼2,4 M olvasatszámot a multiplex szekvenálási futtatás és az egyedi CHIKV-futtatás érte el a hosszabb futási idő miatt (Kiegészítő S1 táblázat). Az egyedi CHIKV- és DENV-szekvenálási futtatásokban az adatok 96%-át, a FLU-A minta esetében pedig 78%-át a spiccelt vírusból származó leolvasások tették ki (1. táblázat). A multiplex szekvenálási adatokban a vírusos leolvasások aránya az egyes vonalkódolt mintákon belül közel áll az egyedileg lefuttatott minták adataihoz (2. táblázat). Mindegyik vírusgenom ultra-magas (>8000) átlagos lefedettségi mélységgel rendelkezett az egyedi és a multiplex szekvenálási adatokban, és a de novo assembly mindhárom vírus esetében 99 közel teljes genomot tudott visszanyerni.9%-os azonosságot a GenBank-referenciához képest.

TABLLE 1. Az egyes könyvtárak MinION szekvenálásából származó adatok térképezési és de novo assembly eredményeinek összefoglalása.

TABLE 2. táblázat. A multiplex MinION szekvenálásból származó adatok térképezési és de novo assembly eredményeinek összefoglalása.
A keresztmintás szennyeződés mértéke és forrása
Minden mintát vonalkóddal láttunk el, és mind egyenként, mind multiplexen szekvenáltuk, ami lehetővé tette, hogy megvizsgáljuk az Albacore vonalkódos demultiplexelésének teljesítményét. Az egyedileg szekvenált minták adataiban csak egyetlen natív vonalkód jelenlétére számítanánk. A CHIKV (NB01 vonalkód), DENV (NB09) és FLU-A (NB10) egyedi szekvenálási futtatások esetében azt találtuk, hogy 86, 109, illetve 17 leolvasást rendeltünk olyan vonalkódtartományokhoz, amelyeknek nem várható a könyvtárban való jelenléte (ami az összes leolvasás 0,0036, 0,0129, illetve 0,001%-át jelenti). A multiplex szekvenálási adatokban 41 leolvasás (0,0016%) olyan vonalkódokhoz volt rendelve, amelyek nem szerepeltek a kísérletekben (azaz nem NB01, NB05, NB09 vagy NB10 vonalkód). Ezeket tévesen hozzárendelt leolvasásokként definiáltuk (2A ábra).
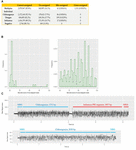
2. ÁBRA. (A) A helyesen hozzárendelt, nem hozzárendelt, rosszul hozzárendelt és kereszthelyzetben hozzárendelt olvasatok számának és százalékos arányának összefoglalása az egyes szekvenálási futtatásokban. A nem hozzárendelt olvasatok az Albacore által 60-nál kisebb vonalkód pontszám miatt egyik tárolóhoz sem rendelhető olvasatokra utalnak, a tévesen hozzárendelt olvasatok azokra az olvasatokra utalnak, amelyeket az ebben a kísérletben nem szereplő tárolókhoz rendeltek, a keresztbe rendelt olvasatok pedig a helytelen vonalkód tárolókhoz rendelt olvasatokra utalnak; (B) az Albacore által a multiplex szekvenálási adatokban a tévesen hozzárendelt olvasatok és a keresztbe rendelt olvasatok esetében bejelentett vonalkód pontszámok eloszlása; (C) egy kiméra és egy helyesen hozzárendelt olvasat nyers jelének összehasonlítása. A kimérikus leolvasás jele egy megakadásjelet és egy hatalmas csúcsjelet tartalmaz a leolvasás közepén.
A szekvenáló könyvtárkészítés során bekövetkező esetleges laboratóriumi szennyeződés vizsgálatához minden egyes futtatásból származó összes leolvasást mindhárom vírus genomiális szekvenciájához illesztettük. Egyetlen olyan olvasatot sem találtunk, amely egy másik könyvtárban készített genomból származott volna, ami arra utal, hogy nem történt in vitro szennyeződés. A multiplex szekvenáló könyvtárat az egyes, nem szennyezett könyvtárak összevonásával állítottuk elő a vonalkód és az adapter ligálása után. A térképezési eredmények azonban azt mutatják, hogy 1311 (0,0543%) olvasat térképeződött a helytelen célgenomhoz, ami arra utal, hogy ezek a leolvasások a rossz vonalkód tárolókhoz lettek kereszthelyezve (a későbbiekben “kereszthelyezett olvasatoknak” nevezzük őket), annak ellenére, hogy a multiplex szekvenáló könyvtárat összevonták az egyedi könyvtárakkal, amelyek egyáltalán nem mutattak kereszthelyezett olvasatokat. Feltételeztük, hogy a tévesen és keresztbe rendelt olvasatok az alacsony vonalkód-pontszám miatt következtek be, és megvizsgáltuk ezen olvasatok vonalkód-pontszámát. A tévesen hozzárendelt olvasatok többsége <70 vonalkód pontszámmal rendelkezett, azonban a kereszt-hozzárendelt olvasatok pontszámai változatosabbak voltak, 60-tól közel 100-ig terjedtek (2B ábra). Ez az eredmény azt sugallta, hogy a rosszul és a keresztbe rendelt olvasatok különböző forrásokból származnak. A kereszthivatkozású leolvasásokat egy kis adatbázissal összevetettük, amely az ebben a vizsgálatban szereplő három vírus genomiális szekvenciáit tartalmazza, és kimutattuk, hogy 1074/1311 (82%) leolvasásuk több vírusgenomhoz (1047 leolvasás) vagy ugyanazon genomon belül különböző régiókhoz (27 leolvasás) volt kereszthivatkozással igazítható, ami arra utal, hogy kimérákról van szó. E megfigyelés megerősítése érdekében megvizsgáltuk néhány kereszt-asszignált olvasat nyers áramjelét a helyesen hozzárendelt olvasatokéval összehasonlítva (2C. ábra). A helyesen hozzárendelt leolvasások aktuális jelei általában a következőket tartalmazzák: (i) egy magas áramerősségű nyitott pórusjelet, amely azt az időt jelzi, amikor a szekvenáló pórus egyik adapterről a másikra vált, (ii) egy stall jelet, amely arra az időszakra utal, amikor a DNS-szekvencia a pórusban van, de még nem mozdult el, és (iii) a DNS-szekvenálás jelnyomát. Ezzel szemben a kiméra leolvasás rendelkezik egy elakadási jellel és egy hatalmas tüskejellel a leolvasás közepén. A kiméra leolvasások két különböző vonalkódszekvenciával rendelkezhetnek az elején és a végén, ami megzavarja a vonalkódtartály hozzárendelését. Ezek az adatok együttesen két hibakategóriát mutatnak, amelyek hozzájárulnak a keresztminta-szennyeződéshez az adatállományunkban: (i) a kimérikus olvasatok (az összes keresztbesorolt olvasat ∼80%-át teszik ki); (ii) az alacsony vonalkód pontszámú olvasatok. A végleges adathalmazunk minőségének javítása érdekében megvizsgáltuk a különböző vonalkód-demultiplexelési megközelítések hatását a kereszthivatkozású olvasatok eltávolítására (3. táblázat). A belső adapterrel rendelkező leolvasások szűrésével a kereszthivatkozású leolvasások 90%-a eltávolítható, és az összes leolvasás 24%-a elveszett. Kipróbáltunk egy szigorúbb szűrési sémát is, amely két vonalkódot követelt meg (egyet-egyet a leolvasás elején és végén) a hozzárendeléshez. Ez a megközelítés két kereszt-asszociált olvasat kivételével minden olvasatot eltávolított, de az összes olvasat 56%-át elvesztette.

TABLÁZAT 3. A kereszthivatkozású olvasatok eltávolítása és a teljes szekvenálási adatvesztés két szűrési megközelítéssel a Porechop segítségével.
Vizsgáljuk a potenciális kiméra olvasatok mértékét is a szekvenálási adatokban. A CHIKV, DENV és FLU-A egyedi szekvenálási futtatások esetében a leképezési eredmények azt mutatják, hogy a leképezett olvasatok 2,3, 3,0 és 2,7%-a rendelkezik kiegészítő igazítással, és legalább kétszer igazodott ugyanahhoz a genomhoz (4. táblázat). A multiplex szekvenálási adatokban mind a vonalkóddal osztályozott, mind a nem osztályozott leolvasásokat figyelembe vesszük. Az eredmények azt mutatják, hogy a leképezett olvasatok 2,0%-a rendelkezik kiegészítő igazítással és legalább kétszer igazodott ugyanahhoz a genomhoz, míg az összes olvasat 0,052%-a legalább két különböző genomhoz igazodott.

TABLE 4. TÁBLA. Összefoglaló a nem-kimerikus, ön-kimerikus és kereszt-kimerikus olvasatok számáról és százalékos arányáról az egyes szekvenálási futtatásokban.
Megbeszélés
Kutatásunk végső célja egy nanopórusos metagenomikai szekvenáláson alapuló diagnosztikai teszt kifejlesztése, amely lehetővé teszi a fertőző betegségek point-of-care vizsgálatát. A multiplex szekvenálás lehetőséget kínál a skálázhatóság javítására és a költségek csökkentésére, azonban a keresztmintaszennyeződés hibákat okozhat az adatokban és az eredmények téves értelmezéséhez vezethet.
Ebben a kísérletben tiszta könyvtárakat egyesítettünk és multiplex MinION szekvenálást végeztünk annak érdekében, hogy megvizsgáljuk a keresztmintaszennyeződés mértékét és forrását. Megállapítottuk, hogy az összes leolvasás 0,056%-a volt keresztsorolva a helytelen vonalkód tárolókhoz, ami hasonló az Illumina szekvenáló platformok esetében különböző tanulmányokból jelentett értékekhez (0,06 és 0,25% között) (Nelson és mtsai., 2014; D’Amore és mtsai., 2016; Wright és Vetsigian, 2016). Eredményeink azt mutatták, hogy a keresztsávkód hozzárendelési hibák domináns forrása a kiméra olvasatok. A kereszthivatkozású kiméra olvasatok ebben az adathalmazban csakis a szekvenálás, nem pedig a könyvtárkészítés során alakulhattak ki, mivel az egyes könyvtárak szekvenálási adataiból teljesen hiányoztak, és az egyetlen további feldolgozási lépés a végleges szekvenáló könyvtárak betöltés előtti keverése volt. Feltételezésünk szerint az Albacore-ban implementált jelenlegi algoritmus nem képes felismerni a nanopóruson egyidejűleg áthaladó DNS-szekvenciák közötti rövid disszociációt, ezáltal több szekvenciát is egy Fast5 fájlba kapcsolva.”
Chimerikus olvasásokat figyeltek meg a MinION szekvenálási adatokban korábban White és munkatársai (2017). Három különböző interferon-amplikon MinION-szekvenálási adatainak elemzése révén a szerzők azt találták, hogy a leképezett olvasatok 1,7%-a kiméra volt. Eredményeink kiegészítik azokat az ismereteket, amelyek alátámasztják, hogy a kimérák gyakoriak a MinION szekvenálási adatokban. Három egyéni és egy multiplex szekvenálási adatban az összes leolvasás 2-3%-a kiméra. Tanulmányunk a következő két szempontból különbözik a korábbi munkáktól. Először is, közvetlen bizonyítékot szolgáltatunk arra, hogy kimérikus olvasatok képződhetnek a könyvtárkészítés után és a szekvenálás során; ezeket a kimérákat továbbá a fent tárgyalt módon a multiplex MinION-szekvenálás során a keresztminták szennyeződésével hoztuk összefüggésbe. Másrészt a kísérleti elrendezésünk korlátozza a könyvtárkészítés során, különösen a standard multiplex szekvenálási protokollban az adaptor ligálás lépése során kialakuló potenciális kimérák azonosítását. Másodszor, eredményeink a MinION-szekvenálás jelenlegi állapotát tükrözik, mivel újabb és legreprezentatívabb ONT-szekvenáló kitet használtunk, beleértve a ligációs szekvenáló kit 1D-t (SQK-LSK108) és a natív barcoding kit 1D-t (EXP-93 NBD103). A nanopórus szekvenálási technológia gyors fejlődés alatt áll, és minden szempontból javulás tapasztalható. Például újabb DNS ligációs szekvenáló készlet (SQK-LSK109) és közvetlen RNS-szekvenáló készlet (SQK-RNA001) jelent meg; az Albacore és Guppy bázishívó algoritmust továbbfejlesztették. Mindezek a változtatások hatással vannak a Nanopore szekvenálási adatokban a kiméra mértékére és a multiplex szekvenálás során a kereszt-barcode szennyeződésre. E tanulmány korlátja a kis számú kísérlet volt, további, különböző kísérleti elrendezéseket alkalmazó munkák hozzájárulnának a Nanopore multiplex szekvenálási adatok megértéséhez. Emellett fontos lenne megvizsgálni a lehetséges tényezők hozzájárulását a kereszt-vonalkód szennyezéshez, ami fényt derítene a multiplex szekvenálási adatok elemzésének legjobb gyakorlatára.
Összefoglalva, tanulmányunk kimutatta, hogy a kimérikus olvasatok a kereszt-vonalkód hozzárendelési hibák domináns forrása a multiplex MinION szekvenálás során. Rávilágít a multiplex MinION-szekvenálási adatok gondos szűrésének szükségességére a downstream elemzés előtt, valamint az érzékenység és a specificitás közötti kompromisszumra, amely a vonalkód-demultiplexelési módszerekre vonatkozik.
A szerzők hozzájárulása
SP, KL, SL és YX végezte a MinION-szekvenálást. YX elemezte az adatokat. Minden szerző megtervezte a vizsgálatot, részt vett az eredmények értelmezésében és a kézirat megírásában, valamint elolvasta és jóváhagyta a kézirat végleges változatát.
Finanszírozás
Ezt a munkát a NIHR Oxford Biomedical Research Centre támogatta.
Érdekütközéssel kapcsolatos nyilatkozat
A szerzők kijelentik, hogy a kutatást olyan kereskedelmi vagy pénzügyi kapcsolatok hiányában végezték, amelyek potenciális összeférhetetlenségként értelmezhetők.
Köszönet
Köszönjük Dr. Anthony Marriottnak (Public Health England) a görény orrspirátumok rendelkezésre bocsátását.