- Introduzione
- Materiali e metodi
- Preparazione del campione
- Preparazione della libreria MinION e sequenziamento
- Analisi genomica
- Risultati
- Dati di sequenziamento MinION e assemblaggio di genomi virali
- Estensione e fonte della contaminazione incrociata dei campioni
- Discussione
- Contributi degli autori
- Finanziamento
- Dichiarazione di conflitto di interessi
- Riconoscimenti
- Materiale supplementare
- Note di commento
Introduzione
Il sequenziamento metagenomico ha il potenziale per consentire l’identificazione imparziale degli agenti patogeni da un campione clinico. Ha la promessa di servire come saggio unico e universale per la diagnostica delle malattie infettive direttamente dai campioni senza la necessità di una conoscenza a priori (Bibby, 2013; Miller et al., 2013; Schlaberg et al., 2017). Oltre all’identificazione delle specie patogene, i dati di sequenza metagenomica ampia e profonda potrebbero fornire informazioni rilevanti per determinare il trattamento e la prognosi, rilevare i focolai e tracciare l’epidemiologia delle infezioni (Greninger et al., 2010; Yang et al., 2011; Qin et al., 2012; Loman et al., 2013). Le piattaforme di sequenziamento di nuova generazione (NGS) possono produrre una massiccia quantità di dati ad un costo modesto, tuttavia, la sua applicazione nella diagnostica clinica e nella sanità pubblica è stata limitata dalla complessità, dalla lentezza e dall’investimento di capitale.
Il MinION è un sequenziatore di genoma a singola molecola in tempo reale, di dimensioni palmari, sviluppato da Oxford Nanopore Technologies (ONT). Le dimensioni compatte e la natura in tempo reale del MinION potrebbero facilitare l’applicazione del sequenziamento metagenomico nei test point-of-care per le malattie infettive, come dimostrato da diversi studi proof-of-concept, tra cui l’identificazione di Chikungunya (CHIKV), Ebola (EBOV), e virus dell’epatite C (HCV) da campioni di sangue clinico umano senza arricchimento del target (Greninger et al, 2015), e il rilevamento di patogeni batterici da campioni di urina (Schmidt et al., 2016) e campioni respiratori, senza la necessità di una coltura preliminare (Pendleton et al., 2017).
Il throughput dei dati di MinION è notevolmente aumentato dal suo rilascio nel 2015, con ogni cella a flusso consumabile che ora genera fino a 10-20 Gb di dati di sequenza del DNA. Questo permette agli utenti di fare un uso più efficiente della cella a flusso (e ridurre i costi) multiplexando diversi campioni in una singola corsa di sequenziamento. ONT ha sviluppato set di codici a barre privi di PCR che consentono il multiplexing di un massimo di 12 campioni.
Il rilevamento del virus dell’influenza A in più campioni respiratori potrebbe essere un uso diagnostico di un saggio di sequenziamento MinION multiplexato. Tuttavia, quando il sequenziamento direttamente da campioni con una potenziale vasta gamma di titoli virali, è importante essere consapevoli del potenziale di contaminazione trasversale del campione, sia durante la preparazione della libreria e la fase di demultiplexing barcode bioinformatica dopo il sequenziamento. Qui, presentiamo un unico set di dati di sequenziamento MinION e i risultati di un’indagine sull’entità e la fonte della contaminazione incrociata dei codici a barre nel sequenziamento multiplex.
Materiali e metodi
Abbiamo usato un campione di lavaggio nasale di furetto infettato dal virus dell’influenza A come esemplare e abbiamo anche aggiunto due aliquote di campioni negativi di lavaggio nasale da furetto non infetto (stock preesistenti non utilizzati da uno studio non correlato) con virus della dengue e del chikungunya separatamente. Nessuno di questi virus è rilevante per la diagnostica clinica in campioni respiratori, ma agiscono qui come marcatori chiari e distinti per la valutazione della contaminazione trasversale del campione. Le librerie di sequenziamento per ogni campione sono state preparate in parallelo, insieme a un controllo negativo del lavaggio nasale, con codice a barre e sequenziate individualmente. Abbiamo poi raggruppato un’aliquota delle librerie di sequenziamento ed eseguito il sequenziamento MinION multiplex. Letture dalle quattro corse individuali (indicato come “CHIKV,” “DENV,” “FLU-A,” e “Negativo”) e la corsa multiplex (indicato come “Multiplexed”) sono stati poi analizzati per indagare l’entità e la fonte di contaminazione del campione croce.
Preparazione del campione
La licenza del progetto è stato esaminato dal locale AWERB (Animal Welfare and Ethics Review Board) ed è stato successivamente concesso dal Ministero degli Interni. L’RNA è stato estratto, utilizzando il kit QIAamp per l’RNA virale (Qiagen) secondo le istruzioni del produttore, dal lavaggio nasale dei furetti contenente il virus dell’influenza A (H1N1) (A/California/04/2009) e da un pool di campioni di lavaggio nasale negativi. Aliquote di estratto del campione negativo sono stati spiked con dengue (DENV) (ceppo TC861HA, GenBank: MF576311) o CHIKV (ceppo S27, GenBank: MF580946.1) RNA virale da La collezione nazionale di virus patogeni1. I campioni sono stati trattati con DNasi utilizzando TURBO DNase (Thermo Fisher Scientific, Waltham, MA, Stati Uniti) e purificati utilizzando il kit RNA Clean & ConcentratorTM-5 (Zymo Research). cDNA è stato preparato e amplificato utilizzando un Sequence-Independent-Single-Primer-Amplification metodi (Greninger et al., 2015) modificato come descritto precedentemente (Atkinson et al., 2016). Il cDNA amplificato è stato quantificato utilizzando il Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA, Stati Uniti), e 1 μg è stato utilizzato come input per ogni preparazione libreria MinION, con l’eccezione del controllo negativo dove l’intero campione (32 ng) è stato utilizzato.
Preparazione della libreria MinION e sequenziamento
Ligazione Sequencing Kit 1D (SQK-LSK108) e Native Barcoding Kit 1D (EXP-NBD103) sono stati utilizzati secondo i protocolli standard ONT, con l’eccezione che solo un codice a barre è stato incluso in ciascuna delle quattro preparazioni libreria. Ogni libreria è stato eseguito su una cella di flusso individuale e una quinta libreria pooled è stato fatto combinando le quattro librerie barcode individualmente. Le librerie sono state sequenziate su celle a flusso R9.4. Il disegno dello studio è mostrato nella Figura 1.
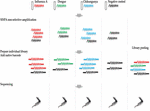
FIGURA 1. Panoramica del disegno dello studio. RNA è stato estratto da quattro campioni, tra cui un campione di lavaggio nasale furetto infettato con il virus dell’influenza A, due campioni negativi furetto lavaggio nasale spikeed con virus dengue e chikungunya, e un controllo negativo furetto lavaggio nasale. cDNA è stato preparato e amplificato utilizzando un Sequence-Independent-Single-Primer-Amplification metodi. Le librerie di sequenziamento per ogni campione sono state preparate in parallelo, con codice a barre e sequenziate su singole celle di flusso. Il sequenziamento multiplex è stato eseguito anche mettendo in comune le quattro librerie individuali. Le letture dalle quattro corse individuali e dalla corsa multiplex sono state analizzate per valutare l’entità e la fonte della contaminazione incrociata dei codici a barre nel sequenziamento multiplex.
Analisi genomica
Le letture sono state basecalled utilizzando Albacore v2.1.7 (ONT) con demultiplexing dei codici a barre. Le letture di ogni corsa di sequenziamento sono state mappate alle sequenze genomiche di ogni virus usando Minimap2 (Li, 2018). Il numero di letture mappate al riferimento è stato contato usando Pysam2. L’assemblaggio de novo è stato eseguito utilizzando Canu v1.7 (Koren et al., 2017), e la bozza di genoma risultante è stata lucidata utilizzando Nanopolish (Mongan et al., 2015) con i dati a livello di segnale.
Per consentire il demultiplexing rigoroso dei codici a barre dei dati di sequenziamento MinION multiplex, abbiamo eseguito due cicli di analisi utilizzando Porechop (v0.2.23). La presenza di una sequenza adattatore nel mezzo di una lettura è una firma di chimera. Abbiamo usato Porechop per esaminare ogni lettura e quelle che hanno la regione centrale che condivide > il 75% di identità con la sequenza adattatore sono state identificate come letture chimeriche. In Porechop, abbiamo impostato l’opzione “-middle_threshold” e scelto una soglia di 75. Nel secondo turno, abbiamo usato Porechop per cercare la sequenza del codice a barre sia all’inizio che alla fine di una lettura; le letture sono state assegnate solo se lo stesso codice a barre è stato trovato alle due estremità. Abbiamo impostato l’opzione “-require_two_barcodes” in Porechop e impostato la soglia per il punteggio del codice a barre a 70. Per trovare la potenziale firma delle letture chimeriche, abbiamo esaminato i segnali di corrente letti memorizzati nel file FAST5 dal sequenziatore MinION. I segnali di corrente sono stati estratti utilizzando ONT fast5 API4 e tracciati utilizzando ggplot2 implementato in R5 per un confronto tra letture chimeriche e non chimeriche.
Risultati
Il throughput di ogni esecuzione di sequenziamento MinION variava a causa delle differenze nel tempo di esecuzione. Un numero massimo di ∼2.4 M legge è stato raggiunto dal sequenziamento multiplexed eseguire e la corsa CHIKV individuale, a causa di tempi di esecuzione più lunghi (Tabella supplementare S1). Letture dal virus spiked rappresentato il 96% dei dati nel CHIKV individuale e DENV sequenziamento corre, e il 78% per il FLU-A campione (Tabella 1). La percentuale di letture virali all’interno di ogni campione con codice a barre nei dati di sequenziamento multiplexed è vicino a quello nei dati del campione eseguito individualmente (Tabella 2). Ogni genoma virale aveva una profondità media di copertura ultra-alta (> 8.000) nei dati di sequenziamento individuale e multiplex, e l’assemblaggio de novo è stato in grado di recuperare genomi quasi completi per tutti e tre i virus con il 99.9% di identità rispetto al riferimento GenBank.

TABLE 1. Riassunto dei risultati di mappatura e assemblaggio de novo per i dati del sequenziamento MinION di singole librerie.

TABILE 2. Riassunto dei risultati di mappatura e di assemblaggio de novo per i dati del sequenziamento MinION multiplex.
Estensione e fonte della contaminazione incrociata dei campioni
Ogni campione è stato dotato di codice a barre e sequenziato sia individualmente che in multiplex, il che ci ha permesso di esaminare le prestazioni del demultiplexing del codice a barre di Albacore. Nei dati del campione sequenziato individualmente ci aspetteremmo che fosse presente solo un singolo codice a barre nativo. Per CHIKV (codice a barre NB01), DENV (NB09), e FLU-A (NB10) corse di sequenziamento individuale, abbiamo trovato che 86, 109, e 17 legge, rispettivamente, sono stati assegnati a bidoni di codici a barre non dovrebbe essere presente nella biblioteca (che rappresentano 0.0036, 0.0129, e 0.001% del totale legge). Nei dati di sequenziamento multiplex, 41 letture (0,0016%) sono state assegnate a codici a barre non inclusi negli esperimenti (cioè, un codice a barre diverso da NB01, NB05, NB09, o NB10). Abbiamo definito queste letture come erroneamente assegnate (Figura 2A).
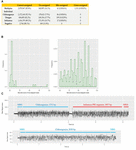
FIGURA 2. (A) riepilogo del numero e della percentuale di letture assegnate correttamente, non assegnate, non assegnate e assegnate in modo incrociato in ogni corsa di sequenziamento. Non assegnato si riferisce a legge che non può essere assegnato a qualsiasi bidone da Albacore a causa di un punteggio codice a barre inferiore a 60, erroneamente assegnato si riferisce a legge che sono stati assegnati a bidoni codice a barre non inclusi in questo esperimento, e cross-assegnato si riferisce a legge che sono stati assegnati al codice a barre errato bidoni; (B) distribuzione dei punteggi codice a barre riportati da Albacore per errato assegnato legge e cross-assegnato legge nei dati di sequenziamento multiplex; (C) confronto del segnale grezzo di un chimerico e una lettura correttamente assegnato. Il segnale della lettura chimerica possiede un segnale di stallo e un enorme segnale di picco al centro della lettura.
Per esaminare la potenziale contaminazione di laboratorio nella preparazione della libreria di sequenziamento, abbiamo mappato tutte le letture di ogni singola corsa rispetto alle sequenze genomiche di tutti e tre i virus. Nessuna lettura è risultata provenire da un genoma preparato in una libreria diversa, suggerendo l’assenza di contaminazione in vitro. La libreria di sequenziamento multiplex è stata preparata unendo le singole librerie non contaminate dopo la legatura del codice a barre e dell’adattatore. Tuttavia, i risultati di mappatura mostrano 1.311 (0,0543%) legge mappato al genoma bersaglio errato, il che implica che sono stati cross-assegnato al barcode errato bins (in seguito denominato “cross-assegnato legge”), nonostante il fatto che la biblioteca di sequenziamento multiplex è stato messo in comune con le librerie individuali non ha mostrato cross-assegnato legge a tutti. Abbiamo ipotizzato che le letture male assegnate e cross-assegnate fossero dovute a un basso punteggio di codice a barre, e abbiamo studiato i punteggi di codice a barre di queste letture. La maggior parte delle letture assegnate erroneamente aveva un punteggio di codice a barre <70, tuttavia, le letture assegnate trasversalmente avevano punteggi più diversi che vanno da 60 a quasi 100 (Figura 2B). Questo risultato ha suggerito che le letture assegnate in modo errato e incrociate provengono da fonti diverse. Abbiamo fatto un blasting delle letture assegnate in modo incrociato con un piccolo database che comprende le sequenze genomiche dei tre virus inclusi in questo studio, e abbiamo dimostrato che 1074/1311 (82%) di queste letture potrebbero essere allineate a più di un genoma virale (1.047 letture) o allineate a regioni distinte all’interno dello stesso genoma (27 letture), suggerendo che sono chimere. Per confermare questa osservazione, abbiamo studiato i segnali di corrente grezzi di alcuni cross-assegnati legge rispetto a quelli di letture correttamente assegnati (Figura 2C). I segnali di corrente di una lettura assegnata correttamente di solito includono: (i) un segnale di poro aperto di alta corrente che rappresenta il tempo che il poro di sequenziamento cambia da un adattatore ad un altro, (ii) un segnale di stallo, riferendosi al periodo di tempo che una sequenza di DNA è nel poro ma ancora a muoversi, e (iii) la traccia del segnale di sequenziamento del DNA. Al contrario, una lettura chimerica possiede un segnale di stallo e un enorme segnale di picco nel mezzo della lettura. Le letture chimeriche possono possedere due diverse sequenze di codice a barre all’inizio e alla fine, confondendo così l’assegnazione di un bin di codice a barre. Presi insieme, questi dati dimostrano due categorie di errore che contribuiscono alla contaminazione trasversale del campione nel nostro set di dati: (i) letture chimeriche (rappresentano il ∼80% di tutte le letture assegnate in modo incrociato); (ii) letture con basso punteggio di codice a barre. Al fine di migliorare la qualità del nostro dataset finale, abbiamo esplorato l’impatto di diversi approcci di demultiplexing del codice a barre per rimuovere le letture cross-assegnate (Tabella 3). Il filtraggio delle letture che possiedono un adattatore interno può rimuovere il 90% delle letture cross-assegnate e perdere il 24% delle letture totali. Abbiamo anche provato uno schema di filtraggio più rigoroso che richiedeva due codici a barre (uno ciascuno all’inizio e alla fine della lettura) per effettuare un’assegnazione. Questo approccio ha rimosso tutte le letture con assegnazioni incrociate tranne due, ma ha perso il 56% delle letture totali.

Tabella 3. Rimozione di cross-assegnato legge e la perdita di dati di sequenziamento totale da due approcci di filtraggio utilizzando Porechop.
Indaghiamo anche la portata di potenziali letture chimeriche nei dati di sequenziamento. Per CHIKV, DENV e FLU-A, i risultati della mappatura mostrano che il 2,3, 3,0 e 2,7% delle letture mappate, rispettivamente, possiede un allineamento supplementare e si allinea almeno due volte allo stesso genoma (Tabella 4). Consideriamo entrambi i codici a barre classificati e non classificati legge nei dati di sequenziamento multiplex. I risultati mostrano che il 2,0% delle letture mappate possiede un allineamento supplementare e si è allineato almeno due volte allo stesso genoma, mentre lo 0,052% delle letture totali è stato allineato ad almeno due genomi distinti.

TABELLA 4. Riassunto del numero e della percentuale di letture non chimeriche, autochimeriche e crosschimeriche in ogni esecuzione di sequenziamento.
Discussione
L’obiettivo finale della nostra ricerca è quello di sviluppare un test diagnostico basato sul sequenziamento metagenomico nanopore che permetta di effettuare test point-of-care per le malattie infettive. Il sequenziamento multiplex offre l’opportunità di migliorare la scalabilità e tagliare i costi, tuttavia, la contaminazione incrociata dei campioni può portare a errori nei dati e alla falsa interpretazione dei risultati.
In questo esperimento, abbiamo messo in comune le librerie pulite ed eseguito il sequenziamento multiplex MinION per indagare l’entità e la fonte della contaminazione incrociata dei codici a barre. Abbiamo identificato lo 0,056% delle letture totali sono state assegnate in modo incrociato ai bidoni di codici a barre errati, che è paragonabile a quelli riportati per le piattaforme di sequenziamento Illumina da diversi studi (tra 0,06 e 0,25%) (Nelson et al., 2014; D’Amore et al., 2016; Wright e Vetsigian, 2016). I nostri risultati hanno mostrato che le letture chimeriche sono la fonte predominante di errori di assegnazione cross-barcode. Le letture chimeriche assegnate in questo set di dati potrebbero essersi formate solo durante il sequenziamento piuttosto che la preparazione della libreria, poiché erano completamente assenti nei dati di sequenziamento delle singole librerie, e l’unico ulteriore passaggio di elaborazione è stato quello di mescolare le librerie di sequenziamento finali prima del caricamento. Ipotizziamo che l’attuale algoritmo implementato in Albacore non possa riconoscere la breve dissociazione tra sequenze di DNA che corrono contemporaneamente attraverso il nanoporo, concatenando così più di una sequenza nello stesso file Fast5.
Le letture chimeriche sono state osservate nei dati di sequenziamento MinION prima in White et al. (2017). Attraverso l’analisi dei dati di sequenziamento MinION di tre diversi ampliconi di interferone, gli autori hanno trovato che l’1,7% delle letture mappate erano chimere. I nostri risultati si aggiungono alle conoscenze a sostegno del fatto che le chimere sono comuni nei dati di sequenziamento MinION. Abbiamo identificato tra il 2 e il 3% delle letture totali in tre dati di sequenziamento individuali e uno multiplex sono chimera. Il nostro studio differisce dal lavoro precedente nei seguenti due aspetti. In primo luogo, forniamo la prova diretta che chimera legge può essere formata dopo la preparazione della libreria e durante il sequenziamento; abbiamo ulteriormente collegato questi chimera alla contaminazione cross-campione in multiplex MinION sequenziamento come discusso sopra. D’altra parte, la nostra configurazione dell’esperimento ha limitazione nell’identificare potenziale chimera formata nella preparazione della biblioteca, particolare durante il passo di legatura adattatore nel protocollo di sequenziamento multiplex standard. In secondo luogo, i nostri risultati riflettono lo stato attuale del sequenziamento MinION perché abbiamo usato più recente e più rappresentativo ONT sequenziamento kit, tra cui legatura sequenziamento kit 1D (SQK-LSK108) e nativo barcoding kit 1D (EXP-93 NBD103). La tecnologia di sequenziamento Nanopore è in rapido sviluppo e il miglioramento avviene in tutti gli aspetti. Per esempio, più recente kit di sequenziamento di legatura del DNA (SQK-LSK109) e kit di sequenziamento diretto RNA (SQK-RNA001) sono stati rilasciati; algoritmo basecalling implementato in Albacore e Guppy basecaller è stato aggiornato. Tutti questi cambiamenti hanno effetto sull’entità della chimera nei dati di sequenziamento Nanopore e sulla contaminazione cross-barcode durante il sequenziamento multiplex. La limitazione di questo studio è stato il piccolo numero di esperimento, lavoro aggiuntivo utilizzando diverse configurazioni di esperimento aggiungerebbe alla nostra comprensione dei dati di sequenziamento multiplex Nanopore. Inoltre, è importante indagare i contributi di potenziali fattori di contaminazione cross-barcode, che avrebbe fatto luce sulle migliori pratiche per analizzare i dati di sequenziamento multiplex.
In sintesi, il nostro studio ha dimostrato che le letture chimeriche sono la fonte predominante di errori di assegnazione cross barcode in multiplex MinION sequenziamento. Evidenzia la necessità di un attento filtraggio dei dati di sequenziamento MinION multiplex prima dell’analisi a valle, e il trade-off tra sensibilità e specificità che si applica ai metodi di demultiplexing dei codici a barre.
Contributi degli autori
SP, KL, SL, e YX hanno condotto il sequenziamento MinION. YX ha analizzato i dati. Tutti gli autori hanno progettato lo studio, hanno partecipato all’interpretazione dei risultati e alla stesura del manoscritto, e hanno letto e approvato la versione finale di questo manoscritto.
Finanziamento
Questo lavoro è stato sostenuto dal NIHR Oxford Biomedical Research Centre.
Dichiarazione di conflitto di interessi
Gli autori dichiarano che la ricerca è stata condotta in assenza di relazioni commerciali o finanziarie che potrebbero essere interpretate come un potenziale conflitto di interessi.
Riconoscimenti
Vorremmo ringraziare il Dr. Anthony Marriott (Public Health England) per aver fornito aspirati nasali di furetto.