Molti dei metodi per visualizzare e interpretare i dati di espressione genica possono essere usati sia per gli esperimenti microarray che per quelli RNA-seq. Alcuni dei metodi più comuni sono discussi di seguito.
Heatmap e clustering
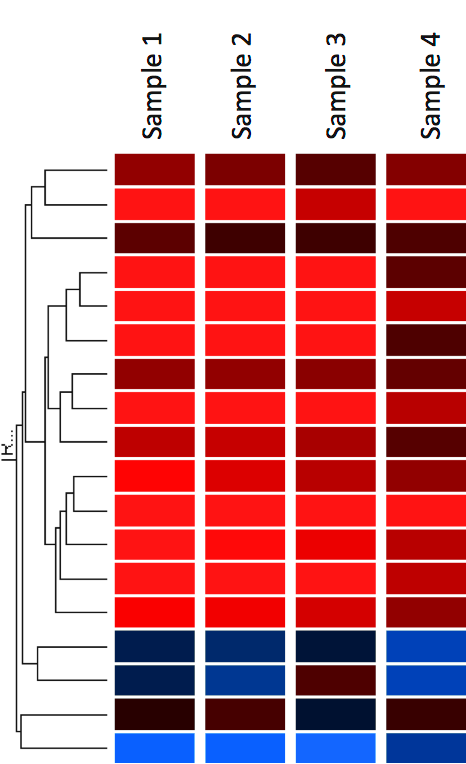
Un metodo comune per visualizzare i dati di espressione genica è quello di visualizzarli come heatmap (Figura 12). La heatmap può anche essere combinata con metodi di clustering che raggruppano i geni e/o i campioni in base alla somiglianza del loro modello di espressione genica. Questo può essere utile per identificare i geni che sono comunemente regolati, o le firme biologiche associate a una particolare condizione (ad esempio una malattia o una condizione ambientale) (4).
Nelle mappe di calore i dati vengono visualizzati in una griglia dove ogni riga rappresenta un gene e ogni colonna rappresenta un campione. Il colore e l’intensità delle caselle è usato per rappresentare i cambiamenti (non i valori assoluti) dell’espressione genica. Nell’esempio qui sotto, il rosso rappresenta i geni up-regolati e il blu i geni down-regolati. Il nero rappresenta l’espressione invariata.
Analisi dell’arricchimento dei set di geni e analisi dei percorsi
Un approccio comune all’interpretazione dei dati di espressione genica è l’analisi dell’arricchimento dei set di geni basata sull’annotazione funzionale dei geni differenzialmente espressi (Figura 13). Questo è utile per scoprire se i geni differenzialmente espressi sono associati ad un certo processo biologico o ad una funzione molecolare.
La Gene Ontology, che contiene l’annotazione standardizzata dei prodotti genici, è comunemente usata per questo scopo. Funziona confrontando la frequenza delle singole annotazioni nella lista dei geni (ad esempio i geni differenzialmente espressi) con una lista di riferimento (di solito tutti i geni sul microarray o nel genoma). L’arricchimento dei percorsi biologici forniti da KEGG, Ingenuity, Reactome o WikiPathways può essere eseguito in modo simile (12,13).
Gli strumenti popolari per l’arricchimento dei set di geni e l’analisi dei percorsi includono:
- DAVID (strumento online gratuito)
- GSEA (gratuito)
- Ingenuity (licenza richiesta)
- Reactome (gratuito)

Analisi della rete
L’analisi della rete è complementare all’analisi del percorso e può essere usata per mostrare come i componenti chiave di diversi percorsi interagiscono. Questo può essere utile per identificare eventi regolatori che influenzano più processi biologici e percorsi (12,13).