INTRODUZIONE
Le malattie polmonari interstiziali formano un gruppo estremamente eterogeneo di disturbi che hanno in comune l’esistenza di una componente infiammatoria del parenchima polmonare che può evolvere in fibrosi e che causa, dal punto di vista funzionale, un’alterazione restrittiva che può portare all’insufficienza respiratoria.
Anche se si tratta di malattie la cui diagnosi e gestione viene effettuata nel campo specialistico della pneumologia, il trattamento viene fondamentalmente seguito su base ambulatoriale, e dato che questo può essere e spesso è punteggiato da varie complicazioni, è importante che il medico di base sia a conoscenza delle attuali linee guida per il trattamento di queste malattie, poiché in molte occasioni sarà consultato da pazienti con questo tipo di problema, sia per complicazioni respiratorie che per altri tipi di complicazioni.
Lo scopo di questa revisione è di aggiornare le modalità terapeutiche che possono essere offerte a questi pazienti oggi. Crediamo che sia della massima importanza non solo per il medico di base avere una conoscenza aggiornata di questo argomento, ma anche per collaborare con lo pneumologo al fine di ottenere una buona conformità con il trattamento del paziente e la diagnosi precoce e il trattamento di eventuali complicazioni che possono sorgere.
Tra le malattie interstiziali, la più rilevante per la sua frequenza e gravità è la fibrosi polmonare idiopatica (IPF). D’altra parte, il suo trattamento è il modello su cui si basa il trattamento di quasi tutte queste malattie, ad eccezione di quelle in cui si può trovare un agente eziologico noto, poiché in queste l’eliminazione di questo agente sarà la base fondamentale della terapia. Ci concentreremo quindi su una revisione delle attuali linee guida per la gestione dell’IPF. Rivedremo prima brevemente le sue caratteristiche cliniche e poi svilupperemo l’attuale gestione dell’IPF in dettaglio.
DESCRIZIONE CLINICA
IPF rappresenta probabilmente il prototipo più aggressivo delle pneumopatie interstiziali diffuse. La sua prevalenza è stata stimata da alcuni autori a 5 casi per 100.000 abitanti1, anche se in realtà è sconosciuta. La sua patogenesi è legata ad alterazioni nei complessi percorsi del sistema immunitario2,3. 2,3 Per questo motivo, la maggior parte dei farmaci utilizzati sono agenti immunosoppressori.
La presentazione clinica più comune è la dispnea ad insorgenza insidiosa accompagnata da una tosse secca, che può essere il sintomo più fastidioso per il paziente a causa della sua natura parossistica e senza respiro. Generalmente, la dispnea è inizialmente irrilevante e un alto indice di sospetto è necessario per una diagnosi precoce. Occasionalmente, il decorso è rapido dall’esordio e porta in pochi mesi all’insufficienza respiratoria irreversibile, come era caratteristico delle descrizioni iniziali di Hamman e Rich. Tuttavia, più comunemente, la condizione è presente da mesi quando il paziente fa la sua prima visita.
All’esame fisico, crepitii fini e secchi (velcro rales) sono molto caratteristici e possono essere sentiti in più dell’80% dei casi. Nei casi avanzati, si può osservare dispnea in caso di lieve sforzo o anche cianosi labiale. Le acropazie possono essere viste in circa il 30-50% dei casi. Il sospetto di IPF è rafforzato quando segni come il modello reticolare, aree di infiltrazione di vetro smerigliato e diminuzione del volume polmonare appaiono sulla radiografia del torace e sulla TAC. Infine, l’esame funzionale confermerà l’esistenza di un modello restrittivo e l’analisi dei gas sanguigni arteriosi confermerà l’esistenza dell’ipossiemia. Nei casi dubbi, la dimostrazione dell’ipossiemia allo sforzo è di grande valore.
La conferma definitiva della diagnosi può essere fatta solo dalla biopsia polmonare. Tuttavia, la realtà è che anche in molte delle serie pubblicate predominano i casi senza diagnosi istologica. Recentemente, l’American Thoracic Society (ATS) ha proposto che l’esistenza di tutti questi criteri significa che, in assenza di esposizione a farmaci, elementi ambientali o malattie del tessuto connettivo, una diagnosi di IPF può molto probabilmente essere fatta in assenza di biopsia polmonare4 nei casi in cui la biopsia polmonare sarebbe un rischio inaccettabile.
La polmonite interstiziale abituale (UIP) è il modello istopatologico che identifica questi pazienti4. Quelli con modelli di polmonite interstiziale desquamativa, malattia interstiziale associata a bronchiolite respiratoria, polmonite interstiziale non specifica, polmonite interstiziale linfoide, polmonite interstiziale acuta e bronchiolite obliterante con polmonite organizzante sono considerati entità diverse.
Una volta stabilite la diagnosi e la gravità clinica, la maggior parte dei pazienti richiede un trattamento perché è una malattia mortale senza remissione spontanea. La risposta al trattamento è scarsa. La sopravvivenza a cinque anni è del 50%5,2,6-8. Il trattamento richiede circa 3-6 mesi prima di stabilire l’efficacia. I rapporti sul trattamento di queste malattie sono scarsi: studi terapeutici non controllati che coinvolgono per lo più un piccolo numero di pazienti9.
Nonostante l’inevitabile progressione insidiosa e irreversibile della malattia, il nichilismo terapeutico non è giustificato. Ci sono esempi di fibrosi non trattata con una progressione lenta che porta a una fibrosi irreversibile che non risponde al trattamento o che sviluppa una fase rapidamente progressiva. Tutti probabilmente richiedono un trattamento al momento della presentazione. Tuttavia, i pazienti con controindicazioni al trattamento o quelli senza evidenza di compromissione funzionale possono seguire un programma di follow-up a intervalli di 3-6 mesi. La gestione dei pazienti anziani con istologia predominante di fibrosi e grave compromissione della funzione polmonare è anche difficile. Questi pazienti ricevono un trattamento breve di 3-6 mesi per determinare la reversibilità. Pochi di loro avranno una risposta positiva.
È spesso difficile determinare se il trattamento produce l’effetto desiderato. Il miglioramento soggettivo si verifica spesso (fino al 70% dei pazienti). Il miglioramento oggettivo si verifica nel 20-30%. I parametri più comunemente utilizzati per definire il miglioramento funzionale (oggettivo) sono un aumento della capacità polmonare totale (TLC) o della capacità vitale (VC), un aumento della capacità di diffusione e una riduzione o normalizzazione del declino della SaO2 durante l’esercizio4,10-12.
TREATMENT
Corticosteroidi
I corticosteroidi sono il pilastro del trattamento dell’IPF, anche se il loro successo è piuttosto scarso: una risposta favorevole si verifica in un terzo dei pazienti. Il razionale del loro uso è quello di sopprimere l’alveolite cronica. Si pensa che funzionino sopprimendo la migrazione dei neutrofili e dei linfociti nel polmone, diminuendo la concentrazione degli immunocomplessi, alterando la funzione dei macrofagi alveolari e l’adesione dei neutrofili alle superfici endoteliali.
Non è chiaro perché alcuni pazienti rispondano e altri no. Può essere legato alla presenza di recettori di superficie sulle cellule parenchimali del polmone. Un breve decorso dei sintomi, una lieve compromissione funzionale e radiologica, il sesso femminile e l’età più giovane dei pazienti sembrano essere associati a un risultato migliore.
Tuttavia, nonostante sia il trattamento più utilizzato, la sua performance a lungo termine non è stata confermata da nessuno studio prospettico, in singolo cieco e controllato con placebo.
Izumi et al hanno analizzato la sopravvivenza di 222 pazienti e hanno scoperto che a 10 anni non c’era differenza di sopravvivenza tra coloro che avevano ricevuto steroidi rispetto ai pazienti non trattati13. Tuttavia, quando un paziente con IPF presenta una funzione polmonare compromessa o sintomi che interferiscono con la vita normale, è ragionevole iniziare un tentativo di terapia corticosteroidea. Infatti, il miglioramento spontaneo della malattia è molto raro, e se si decide di ritardare l’inizio del trattamento dovrebbe essere a scapito di uno stretto monitoraggio del paziente in modo che il trattamento possa essere iniziato non appena il deterioramento è evidente.
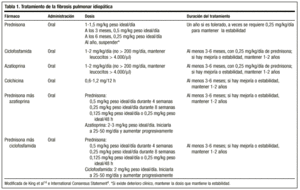
Il trattamento classico inizia con prednisone 1,5-2 mg/kg di peso ideale/giorno come dose singola al mattino, per non superare una dose di 100 mg. Dopo 2-3 mesi, il paziente viene rivalutato con radiografia e studio funzionale. Se il paziente risponde, il prednisone viene diminuito (1-2 mg/settimana) fino a una dose di mantenimento di 0,5 mg/kg di peso ideale/giorno, che può essere ridotta in seguito. Se non c’è risposta, viene aggiunto un farmaco immunosoppressore. In generale, è improbabile che il trattamento possa essere interrotto, e le dosi di 0,25 mg/kg di peso ideale/giorno sono solitamente mantenute.
Di recente, la Società Toracica Americana e quella Europea, per evitare gli effetti collaterali di questo farmaco e per ottenere migliori tassi di sopravvivenza, hanno raggiunto un consenso per iniziare il trattamento con una combinazione di prednisone o un altro corticosteroide a dosi equivalenti e un immunosoppressore (azatioprina o ciclofosfamide)4: prednisone 0,5 mg/kg di peso ideale/giorno per 4 settimane; 0,25 mg/kg di peso ideale/giorno per 8 settimane, poi si passa a 0,125 mg/kg di peso ideale/giorno o 0,25 mg/kg di peso ideale/48 h.
Non ci sono linee guida sulla durata del trattamento; il tempo minimo è un anno. È possibile che dopo mesi o anni la malattia si riattivi, nel qual caso il trattamento con gli stessi agenti dovrebbe essere iniziato. In ogni caso, la risposta al trattamento dovrebbe sempre essere valutata in termini di miglioramento oggettivo, e la possibilità di mantenere il paziente cronicamente su una dose che garantisca la stabilità dovrebbe essere riservata a quei casi in cui c’è un miglioramento con i corticosteroidi, un peggioramento al tapering e un nuovo miglioramento aumentando nuovamente la dose. La dose necessaria per ogni paziente può essere determinata solo caso per caso, a seconda della risposta ai cambiamenti di dose. Molti autori considerano necessario un trattamento per tutta la vita.14
Quando la malattia è inizialmente grave, sono stati proposti corticosteroidi ad alte dosi. Keogh et al hanno usato corticosteroidi parenterali ad alte dosi intermittenti in pazienti con malattia grave: metilprednisolone endovena, 2 g una volta alla settimana, più prednisone orale, 0,25 mg/kg/giorno. La logica è che hanno un effetto più pronunciato sull’alveolite neutrofila (associata a una prognosi sfavorevole) rispetto agli steroidi a basso dosaggio15. Tuttavia, questo lavoro soffre di alcune carenze, come il breve follow-up di 6 mesi, il piccolo numero di pazienti e le dosi di prednisone inferiori allo standard9. Il trattamento iniziale con corticosteroidi ad alte dosi per via endovenosa (metilprednisolone 250 mg/6 h) è stato utilizzato nel tentativo di controllare la malattia il più presto possibile quando è rapidamente progressiva. Non ci sono studi controllati per determinare se questi metodi sono efficaci; tuttavia, dosi più alte sembrano avere un’efficacia maggiore, sebbene gli effetti collaterali siano anche maggiori.14.
AGENTI CITOTOSSICI
Sono stati usati classicamente come alternativa ai corticosteroidi quando questi erano inefficaci, e anche quando i loro effetti collaterali precludono la loro ulteriore somministrazione. In linea con lo spirito eminentemente pratico di questo articolo, passeremo in rassegna gli agenti che si sono dimostrati utili nella pratica clinica, anche se, come nel caso dei corticosteroidi, mancano serie lunghe, prospettiche e randomizzate che ci permettano di sapere esattamente quale sia il loro ruolo esatto nel trattamento della IPF.
Azatioprina
Il suo meccanismo d’azione si basa sulla sostituzione delle purine nella sintesi dell’acido desossiribonucleico, inibendo così l’adenina deaminasi e inattivando i linfociti. Altri effetti includono la soppressione dell’attività delle cellule natural killer, la produzione di anticorpi e la citotossicità cellulare anticorpo-dipendente.
La dose raccomandata è 2 mg/kg/giorno, per non superare i 200 mg/giorno, per un periodo di 3-6 mesi. La somministrazione viene iniziata alla dose di 50 mg/giorno e, se ben tollerata, la dose viene aumentata a intervalli di 1 o 2 settimane fino al raggiungimento della dose prevista.
I principali effetti collaterali sono ematologici: leucopenia, anemia, trombocitopenia, aplasia dei globuli rossi e pancitopenia. È necessario monitorare la conta dei leucociti, e se scende al di sotto di 4.000/μl, l’azatioprina deve essere interrotta fino a quando questa cifra non sale di nuovo. Inoltre, possono verificarsi disturbi gastrointestinali come nausea e vomito, ulcera peptica, diarrea e lieve elevazione degli enzimi epatici. Nel complesso, fino al 20-30% dei casi, gli effetti collaterali richiedono la sospensione dell’azatioprina.
Raghu et al hanno confrontato l’efficacia dei glucocorticoidi ad alte dosi rispetto all’azatioprina in combinazione con glucocorticoidi a basse dosi. L’uso di questo farmaco è stato accompagnato da un modesto miglioramento dei parametri della funzione respiratoria e da un aumento della sopravvivenza17.
Ciclofosfamide
È un agente alchilante. La sua modalità d’azione è la deplezione dei linfociti e quindi sopprime la funzione dei linfociti. La dose raccomandata è di 2 mg/kg/giorno singola, orale, di solito considerata con 0,25 mg/kg/giorno di prednisone orale, per non superare i 200 mg/giorno per almeno 3 mesi e fino a 9-12 mesi.
L’effetto collaterale più importante è la leucopenia. Deve essere mantenuta una conta totale dei leucociti superiore a 3.000/μl o una conta dei neutrofili superiore a 1.500/μl. Altre complicazioni includono trombocitopenia, ematuria secondaria a cistite emorragica, anoressia, nausea e vomito, soppressione del midollo osseo, azoospermia e amenorrea, infezioni e malignità ematologiche.
Kolb et al hanno studiato 18 pazienti con IPF per determinare l’efficacia e la sicurezza della terapia con ciclofosfamide pulsata. La ciclofosfamide è stata somministrata in modo intermittente (1-1,3 g/mese) insieme al prednisolone orale per un anno. Dopo questo periodo, la ciclofosfamide è stata interrotta e le dosi raggiunte di corticosteroidi sono state continuate8 . Sono stati seguiti per un minimo di 14 mesi, monitorando QoL, TLC, FEV1, capacità di trasferimento di CO (DLCO), SaO2, così come i sintomi. Un paziente ha dovuto essere interrotto a causa di polmoniti ricorrenti; 11 pazienti sono stati considerati rispondenti (5 migliorati e 6 stabilizzati) e 6 peggiorati. La tossicità a lungo termine della ciclofosfamide dipende dalle dosi cumulative. Le dosi intermittenti sono altrettanto efficaci e meglio tollerate delle dosi giornaliere. I rispondenti avevano una percentuale più alta di linfociti nel BAL. Un altro risultato degno di nota è che la QOL iniziale dei responders era significativamente migliore a causa di un tempo più breve alla progressione, suggerendo che questo trattamento dovrebbe essere iniziato presto.
Baughman e Lower hanno trattato 33 pazienti IPF con 1.000-1.800 mg di ciclofosfamide/2 settimane per 18 mesi. Coloro che sono sopravvissuti > 18 mesi hanno avuto un miglioramento significativo in VCF che è stato mantenuto nel corso dell’anno successivo. La riduzione del prednisolone è stata possibile da 32 a 4 mg/giorno16.
Penicillamina
Agente antifibrosante. È stato suggerito che interferendo con la formazione di legami covalenti intra- e intermolecolari delle fibre di collagene inibisce lo sviluppo della fibrosi polmonare. Sono stati descritti alcuni casi di malattie del tessuto connettivo con fibrosi polmonare associata in cui ha avuto una certa efficacia, ma non ci sono studi controllati in grandi serie di pazienti, quindi non può essere considerato attualmente come un valido trattamento alternativo. Sono quindi necessari ulteriori studi prima di poter fare una raccomandazione.
Colchicina
Riduce la secrezione di collagene e aumenta l’attività collagenolitica. Ha anche un effetto antinfiammatorio. Sono necessari studi per determinare più precisamente il suo ruolo nel trattamento di questa malattia, ma ci sono studi in cui si attribuisce la sua efficacia essendo simile a quella dei corticoidi, e i suoi effetti collaterali non sono gravi. È raccomandato alla dose di 0,6 mg, una o due volte al giorno, in pazienti refrattari ai corticosteroidi, da soli o in combinazione con agenti immunosoppressivi. Anche se ci sono autori che la collocano nella prima linea di trattamento, attualmente mancano i dati per stabilirla come tale (tabella 1).
Trapianto polmonare
I casi in cui la malattia progredisce senza risposta al trattamento e finisce per provocare insufficienza respiratoria sono, purtroppo, i più numerosi in questa malattia. Non c’è attualmente alcuna prospettiva di trattamento medico per questi pazienti, tranne la palliazione. In questo scenario desolante, la possibilità del trapianto di polmone è emersa negli anni ’80 e si è sviluppata al punto che ora è un’opzione terapeutica che dovrebbe essere considerata come uno strumento di uso clinico e non è più limitata al campo sperimentale. La sua indicazione più adatta è nelle persone sotto i 65 anni di età, in assenza di malattia progressiva e irreversibile in altri organi, così come l’infezione attiva non risolta o l’infezione da germi multi-resistenti. Sono necessari anche un’adeguata predisposizione personale e uno stato nutrizionale. I pazienti con una CPT inferiore al 60% del previsto o una capacità di diffusione polmonare inferiore al 40% hanno una sopravvivenza inferiore a 2 anni. In questo grado di disfunzione richiedono una terapia di ossigeno e si desaturano facilmente con un esercizio minimo. Il trapianto di polmone dovrebbe essere considerato in questi casi, non appena l’ossigenoterapia diventa necessaria e si verifica una progressione inarrestabile della malattia. Il tipo di trapianto di scelta è il trapianto di un polmone, riservando il trapianto di due polmoni ai casi in cui ci sono dubbi sul comportamento del polmone residuo. Quando il paziente ha meno di 50 anni e ha un’insufficienza cardiaca destra, il trapianto cardiopolmonare può essere considerato. Tuttavia, in questa situazione la malattia è molto avanzata ed è improbabile che resista all’attesa18,19. La sopravvivenza post-trapianto dovrebbe essere dell’80% nel primo anno e del 50-60% a 5 anni, il che migliora significativamente la prognosi per un paziente con IPF che ha raggiunto l’insufficienza respiratoria.
VALUTAZIONE DELLA RISPOSTA AL TRATTAMENTO
In assenza di complicazioni, possono essere necessari fino a 6 mesi prima di ottenere una risposta al trattamento. Dopo questo tempo, se il paziente è migliorato o è stabile, le stesse dosi dovrebbero essere mantenute. Se sono peggiorati, devono essere passati a una terapia alternativa (un altro agente immunosoppressivo o anche il trapianto). A 12 mesi, dovrebbe essere rivalutato allo stesso modo e dopo 18 mesi, individualizzato in base alla risposta. Se recomienda continuar indefinidamente sólo en aquellos pacientes con mejoría continua o estabilización (tabla 2).

Una respuesta favorable se define por dos o más de los siguientes criterios en dos visitas consecutivas:
1. Mejoría de los síntomas, sobre todo en el grado de esfuerzo requerido para que el paciente deba parar cuando hace un esfuerzo o un descenso en la frecuencia o gravedad de la tos.
2. Reducción de las alteraciones radiológicas.
3. Mejoría funcional definida por dos o más de los siguientes parámetros:
Aumento >= 10% en la TLC o CV (o >= 200 ml).
Aumento >= 15% en DLCO o >= 3 ml/min/mmHg.
Mejoría o normalización de la SaO2 o pO2 (aumento >= 4% o >= 4 mmHg) durante un prueba de esfuerzo cardiorrespiratoria.
Una respuesta estable (también considerada favorable) se define por dos o más de los siguientes criterios en dos visitas consecutivas:
1. Cambio de
2. Cambio de
3. Sin cambios en la SaO2 o pO2 (aumento
Un fracaso del tratamiento se define por dos o más de los siguientes criterios en dos visitas consecutivas:
1. Aumento de los síntomas, sobre todo disnea o tos.
2. Aumento de las alteraciones radiológicas, especialmente imágenes en panal o signos de hipertensión pulmonar.
3. Deterioro funcional definido por dos o más de los siguientes parámetros:
Descenso >= 10% en la TLC o CV (o >= 200 ml).
Descenso >= 15% en DLCO o >= 3 ml/min/mmHg.
Peggioramento della SaO2 (>= 4%) o aumento del gradiente alveolo-arterioso (AapO2) (>= 4 mmHg) a riposo o durante un test da sforzo cardiorespiratorio.
CONCLUSIONI
La fibrosi polmonare idiopatica è una malattia mortale senza remissione spontanea e con scarsa risposta al trattamento. Gli studi su quest’ultimo sono scarsi e coinvolgono un piccolo numero di pazienti. I corticosteroidi mantenuti per un minimo di un anno a dosi diverse sono il regime più accettato, da solo o in combinazione con altri agenti immunosoppressivi. Recentemente, la terapia combinata è stata raccomandata dall’American Thoracic e dalla European Respiratory Societies4 . Gli agenti anti-fibrosi vengono studiati con l’intenzione di impedire che il processo fibrotico si instauri e diventi quindi irreversibile, dato che molti pazienti vengono diagnosticati in fase avanzata. Dati i tassi di sopravvivenza e l’evoluzione, il trapianto di polmone dovrebbe essere considerato non appena i pazienti falliscono il trattamento e richiedono l’ossigenoterapia. Tutto ciò suggerisce la necessità di stabilire studi multicentrici con l’idea di reclutare un numero sufficiente di pazienti per valutare l’efficacia dei trattamenti somministrati, così come per identificare la storia naturale della malattia e la reale incidenza e prevalenza4,20. D’altra parte, si raccomanda di identificare e iniziare il trattamento nelle prime fasi della malattia per ottenere tassi di sopravvivenza più elevati.