遺伝子発現データを可視化し解釈する方法の多くは、マイクロアレイとRNA-seq実験の両方に使用することが可能である。
ヒートマップとクラスタリング
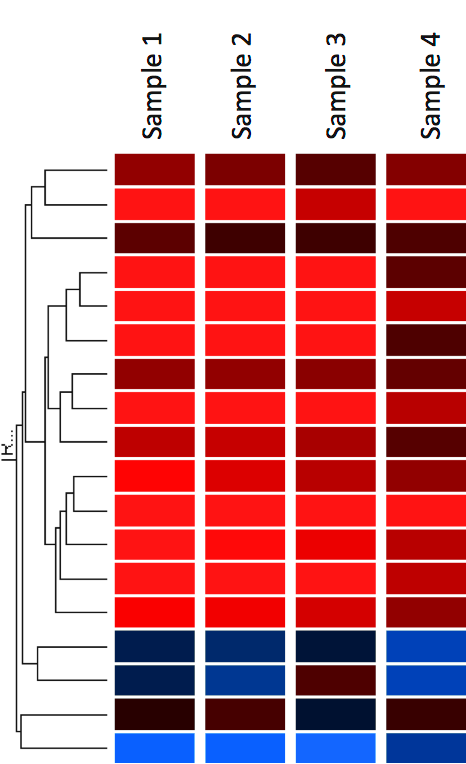
遺伝子発現データを可視化する一般的な方法は、ヒートマップとしてそれを表示することです(図12)。 ヒートマップは、遺伝子発現パターンの類似性に基づいて遺伝子および/またはサンプルをグループ化するクラスタリング手法と組み合わせることもできる。
ヒートマップでは、各行が遺伝子、各列がサンプルを表すグリッドでデータが表示されます。 箱の色と強度は、遺伝子発現の変化(絶対値ではない)を表すために使用されます。 以下の例では、赤が発現量の増加した遺伝子、青が発現量の減少した遺伝子を表しています。
遺伝子セット濃縮解析とパスウェイ解析
遺伝子発現データの解釈でよくあるアプローチは、差動発現遺伝子の機能注釈に基づく遺伝子セット濃縮解析です(図 13)。
遺伝子産物の標準的なアノテーションを含むGene Ontologyは、この目的のために一般的に使用されています。
この目的のためには、遺伝子産物の標準的なアノテーションを含むGene Ontologyがよく使われる。これは、遺伝子リスト中の個々のアノテーション(例えば、差次的に発現した遺伝子)の頻度を参照リスト(通常はマイクロアレイまたはゲノム中のすべての遺伝子)と比較することによって機能する。 KEGG, Ingenuity, Reactome, WikiPathways などで提供されている生物学的パスウェイのエンリッチメントも同様の方法で行うことができる(12,13)。
遺伝子セットのエンリッチメントとパスウェイ解析のための一般的なツールは以下の通りである。
- DAVID(無料オンラインツール)
- GSEA(無料)
- Ingenuity(要ライセンス)
- Reactome(無料)

ネットワーク解析
ネットワーク解析はパスウェイ解析を補完し、異なるパスウェイの主要構成要素がどのように相互作用するかを示すのに用いることができる。 これは、複数の生物学的プロセスやパスウェイに影響を与える制御イベントを特定するのに役立ちます(12,13)
。