はじめに
メタゲノムシーケンスは、臨床サンプルから病原体を偏りなく同定できる可能性を秘めています。 それは、アプリオリな知識を必要とせず、サンプルから直接感染症を診断するための単一かつ普遍的なアッセイとして機能することが期待されている(Bibby, 2013; Miller et al, 2013; Schlaberg et al, 2017)。 病原体種の同定に加えて、広範かつ深いメタゲノム配列データは、治療や予後の決定、アウトブレイクの検出、感染症疫学の追跡に関連する情報を提供しうる(Greningerら、2010年;Yangら、2011年;Qinら、2012年;Lomanら、2013年)。 次世代シーケンサー(NGS)プラットフォームは、控えめなコストで大量のスループットデータを生成できますが、臨床診断や公衆衛生への応用は、複雑さ、遅さ、設備投資によって制限されています。
MinIONは、オックスフォードナノポア・テクノロジー(ONT)が開発した手のひらサイズのリアルタイム1分子ゲノム・シーケンサーです。 MinIONのコンパクトなサイズとリアルタイム性は、いくつかの概念実証研究によって実証されているように、感染症のポイントオブケア検査におけるメタゲノムシーケンスの適用を促進する可能性があり、ターゲット濃縮なしでヒト臨床血液サンプルからチクングニア(CHIKV)、エボラ(EBOV)、C型肝炎ウイルス(HCV)の特定(Greninger et al, 2015)、および尿サンプル(Schmidtら、2016)および呼吸器サンプルから、事前の培養を必要としない細菌病原体の検出(Pendletonら、2017)
2015年のリリース以来、MinIONのデータスループットは大幅に向上し、各消耗フローセルは現在最大10~20 GbのDNAシーケンスデータを生成しています。 これにより、ユーザーは1回のシーケンスランで複数のサンプルをマルチプレックスすることで、フローセルをより効率的に使用することができます(コストも削減できます)。
複数の呼吸器系サンプルにおけるA型インフルエンザウイルスの検出は、多重化MinIONシーケンスアッセイの診断的用途の1つとなりえます。 しかし、幅広いウイルス力価を持つ可能性のあるサンプルから直接シーケンスを行う場合、ライブラリ調製時およびシーケンス後のバイオインフォマティクスバーコードデマルチプレックス段階の両方で、クロスサンプル汚染の可能性を認識することが重要である。
材料と方法
私たちは、A型インフルエンザウイルスに感染したフェレットの鼻洗浄サンプルを模範として使用し、感染していないフェレットの陰性鼻洗浄サンプルのアリコート2個(関連のない研究で使用しなかった既存のストック)に、デングウイルスとチクングニアウイルスも別々に混入させた。 これらのウイルスはいずれも呼吸器系サンプルの臨床診断に関連するものではないが、ここではサンプル間汚染を評価するための明確で異なるマーカーとして機能する。 各サンプルのシーケンスライブラリは、陰性鼻腔洗浄コントロールと並行して調製され、バーコード化され、個別にシーケンスされました。 その後、シーケンスライブラリのアリコートをプールし、マルチプレックスMinIONシーケンスを実施した。
サンプル調製
プロジェクトライセンスは、地元のAWERB (Animal Welfare and Ethics Review Board)によって審査され、その後、Home Officeによって承認されました。 インフルエンザA(H1N1)ウイルス(A/California/04/2009)を含むフェレットの鼻洗浄と、陰性の鼻洗浄サンプルのプールから、QIAampウイルスRNAキット(Qiagen)を使用して、メーカーの説明書に従ってRNAを抽出した。 陰性サンプル抽出液のアリコートに、The National collection of Pathogenic Viruses1 からデング熱(DENV)(TC861HA株、GenBank:MF576311)またはチクビ(S27株、GenBank:MF580946.1)のいずれかのウイルスRNAをスパイクした。 サンプルは、TURBO DNase(Thermo Fisher Scientific, Waltham, MA, United States)を用いてDNase処理し、RNA Clean & ConcentratorTM-5 kit(Zymo Research)を用いて精製した。 cDNAは準備し、以前に記載したように修正した配列-独立-単一-プライマー増幅法(Greninger et al, 2015) (Atkinson et al., 2016)を用いて増幅させた。 増幅されたcDNAは、Qubit dsDNA HS Assay Kit(Thermo Fisher Scientific, Waltham, MA, United States)を用いて定量し、全サンプル(32 ng)を使用した陰性対照を除き、各MinIONライブラリー調製の入力として1 μgを使用した。
MinION Library Preparation and Sequencing
Ligation Sequencing Kit 1D (SQK-LSK108) と Native Barcoding Kit 1D (EXP-NBD103) をONT標準プロトコルに従って使用し、ただし、4つのライブラリ準備にはそれぞれ1つのバーコードのみが含まれるようにしました。 各ライブラリーは個別のフローセルで実行され、5番目のプールライブラリーは4つの個別にバーコード化されたライブラリーを結合して作製された。 ライブラリーはR9.4フローセルでシーケンスされた。 研究デザインを図1に示す。
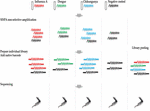
FIGURE 1.研究デザインは、図1.に示すとおりである。 研究デザインの概要。 RNAは、A型インフルエンザウイルスに感染したフェレット鼻腔洗浄サンプル、デングウイルスおよびチクングニアウイルスにスパイクした2つの陰性フェレット鼻腔洗浄サンプル、および陰性フェレット鼻腔洗浄対照を含む4サンプルから抽出された。 各サンプルのシーケンスライブラリーを並行して調製し、バーコードを付けて、個々のフローセルでシーケンスした。 また、4つのライブラリーをプールしてMultiplexシーケンスを行った。
ゲノミクス解析
バーコード除去を行ったAlbacore v2.1.7 (ONT) を用いてリードのベースコールを行った。 各シーケンスランのリードは、Minimap2(Li, 2018)を用いて各ウイルスのゲノム配列にマッピングされた。 Pysam2を用いて、参照にマッピングされたリードの数をカウントした。 De novoアセンブリはCanu v1.7 (Koren et al., 2017)を用いて行い、得られたドラフトゲノムはシグナルレベルデータを用いてNanopolish (Mongan et al., 2015)で研磨した。
多重MinIONシーケンスデータの厳格なバーコードデマルチプレックスを可能にすべく、Porechop (v0.2.23) を用いて2ラウンド分の分析を実行した。 リードの途中にアダプター配列があることは、キメラのサインである。 Porechopを用いて各リードの解析を行い、中間領域がアダプター配列と>75% 同一であるものをキメラリードとして同定しました。 Porechopでは、”-middle_threshold “オプションを設定し、75%の閾値を選択しました。 2回目は、Porechopを用いて、リードの始点と終点の両方でバーコード配列を検索し、両端で同じバーコードが見つかった場合のみ、リードを割り当てた。 Porechopのオプションに”-require_two_barcodes “を設定し、バーコードのスコアの閾値を70に設定した。 キメラリードのシグネチャーを検出するために、MinIONシーケンサーでFAST5ファイルに格納されたリード電流シグナルを調べた。
結果
MinION Sequencing Data and Assembly of Viral Genomes
各MinIONシーケンサーのスループットは、実行時間の違いによりばらつきがあることがわかった。 最大で約2.4Mのリードが、multiplexed sequencing runと個々のCHIKV runで達成されたが、これは実行時間が長かったためである(補足表S1)。 スパイクされたウイルスからのリードは、CHIKVとDENVの個別シーケンスランではデータの96%を占め、FLU-Aサンプルでは78%でした(Table 1)。 Multiplexedシーケンスデータにおける各バーコードサンプル内のウイルスリードの割合は、個別に実行したサンプルデータの割合とほぼ同じです(Table 2)。 各ウイルスゲノムは、個別およびMultiplexシーケンスデータにおいて超高濃度(>8,000) 平均カバー深度を有しており、de novoアセンブリにより、3つのウイルスすべてについてほぼ完全なゲノムを99.0%で復元することができた。

TABLE 1.ゲノム解析

TABLE 2. multiplex MinION sequencingのデータに対するマッピングとde novoアセンブリの結果のまとめ
サンプル間汚染の範囲と原因
それぞれのサンプルはバーコード化され、個別と多重の両方で配列決定され、これによりAlbacoreのバーコードデマルチプレックスの性能を調べることができました。 個別に配列されたサンプルデータでは、単一のネイティブバーコードのみが存在することが予想されます。 CHIKV (barcode NB01), DENV (NB09), FLU-A (NB10) の個別シーケンスランでは、それぞれ86, 109, 17リードがライブラリに存在しないバーコードビンに割り当てられたことがわかりました(総リードの0.0036、 0.0129, 0.001% に相当します)。 マルチプレックスシーケンスデータでは、41リード(0.0016%)が実験に含まれないバーコード(すなわち、NB01、NB05、NB09、NB10以外のバーコード)に割り当てられていました。
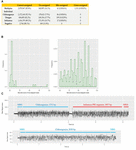
FIGURE 2.FIGURE 2. (A)各シーケンスランで正しく割り当てられたリード、未割り当て、誤割り当て、および交差割り当ての数および割合のまとめ。 (B)マルチプレックスシーケンスデータ中のミスアサインリードとクロスアサインリードについてAlbacoreが報告したバーコードスコアの分布、(C)キメラリードと正しくアサインされたリードの生シグナルの比較。
シーケンスライブラリ作成における実験室での汚染の可能性を調べるために、各個人実行からのすべてのリードを3つのすべてのウイルスのゲノム配列に対してマッピングしました。 別のライブラリで調製されたゲノムに由来するリードは見つからず、in vitroでのコンタミネーションはないことが示唆された。 マルチプレックスシーケンスライブラリーは、バーコードとアダプターの両方をライゲーションした後、汚染されていない個々のライブラリーをプールすることによって調製された。 しかし、マッピングの結果、1,311本(0.0543%)のリードが誤ったターゲットゲノムにマッピングされ、誤ったバーコードビンにクロスアサインされたことが示唆された(後に「クロスアサインリード」と呼ぶ)。しかし、個々のライブラリーをプールして多重化した結果、クロスアサインリードが全く認められなかったにもかかわらずである。 我々は、ミスアサインリードやクロスアサインリードは、バーコードスコアが低いことが原因であると考え、これらのリードのバーコードスコアを調査した。 その結果、ミスアサインリードの多くはバーコードスコア<70 であったが、クロスアサインリードは60から100近くまで多様なスコアを有していた(図2B)。 この結果は、ミスアサインリードとクロスアサインリードが異なるソースに由来することを示唆するものであった。 本研究で対象とした3種類のウイルスのゲノム配列からなる小規模なデータベースにクロスアサインしたところ、1074/1311本(82%)のリードが複数のウイルスゲノムにクロスアラインされるか(1047本)、同じゲノム内の異なる領域にクロスアラインされる(27本)ことが示され、キメラであることが示唆された。 この結果を確認するために、正しく割り当てられたリードと、クロスアラインされたリードの電流シグナルを比較しました(図2C)。 図2C)。正しく割り当てられたリードの電流シグナルは、通常、以下のものを含んでいる。 (i)配列決定孔があるアダプターから別のアダプターに変わる時間を表す高電流の開孔信号、(ii)DNA配列が孔の中にあるがまだ動いていない時間を表す失速信号、(iii)DNA配列決定の信号跡が含まれる。 一方、キメラリードは、ストール信号と途中に大きなスパイク信号がある。 また、キメラリードは、始点と終点に2つの異なるバーコード配列を持つため、バーコードビンの割り当てに混乱をきたす。 これらのデータを総合すると、我々のデータセットにおけるクロスサンプル汚染の原因となる2つのエラーカテゴリーが示される。 (i) キメラリード(全クロスアサインリードの約80%を占める)、(ii) バーコードスコアが低いリード。 最終的なデータセットの品質を向上させるために、クロスアサインリードを除去するための様々なバーコードデマルチプレックス手法の影響を検討した(表3)。 内部アダプターを持つリードのフィルタリングにより、クロスアサインリードの90%を除去でき、全リードの24%を損失した。 さらに、2つのバーコード(リードの始点と終点にそれぞれ1つずつ)を必要とする、より厳格なフィルタリング方式も試みました。

TABLE 3.交差配列リードを除くすべてのリードが失われた。 Porechopを用いた2つのフィルタリングアプローチによるクロスアサインリードと全シーケンスデータの損失の除去
我々はまた、シーケンスデータ中の潜在的キメラリードの程度を調査しています。 CHIKV、DENV、FLU-Aの各シーケンスランについて、マッピングの結果、マッピングされたリードのそれぞれ2.3、3.0、2.7%が補足アライメントを持っており、同じゲノムに少なくとも2回アライメントしていることが分かった(表4)。 マルチプレックスシーケンスデータ中のバーコードで分類されたリードと分類されていないリードの両方を考慮しました。 その結果、マッピングされたリードの2.0%がsupplementary alignmentを持ち、少なくとも2回同じゲノムにアラインメントされることが示された一方、全リードの0.052%が少なくとも2つの異なるゲノムにアラインメントされた。 各シーケンスランにおける非キメラ、セルフキメラ、クロスキメラのリードの数と割合のまとめ。
考察
我々の研究の最終目的は、感染症のポイントオブケア検査を可能にするナノポアメタゲノムシーケンスに基づいた診断アッセイの開発である。
本実験では、クロスバーコード汚染の程度と原因を調べるため、クリーンなライブラリーをプールし、マルチプレックスMinIONシーケンシングを実施しました。 我々は、総リードの0.056%が不正なバーコードビンにクロスアサインされていることを確認したが、これは異なる研究によるイルミナシーケンスプラットフォームの報告(0.06~0.25%)と同等である(Nelsonら、2014; D’Amore ら、2016; Wright and Vetsigian、2016)。 我々の結果は、キメラリードがクロスバーコードアサインエラーの主な原因であることを示した。 このデータセットにおけるクロスアサインキメラリードは、個々のライブラリのシーケンスデータには全く存在せず、さらなる処理ステップはロード前に最終的なシーケンスライブラリを混合することだけであったため、ライブラリ準備ではなくシーケンス中にのみ形成された可能性がある。 Albacoreに実装されている現在のアルゴリズムでは、ナノポアを同時に走るDNA配列間の短い解離を認識できず、それによって複数の配列が同じFast5ファイルに連結されていると仮定しています。
Chimericリードは、Whiteら(2017)において以前にMinIONシーケンスデータで観察されています。 3つの異なるインターフェロンアンプリコンのMinIONシーケンスデータの分析を通じて、著者らはマッピングされたリードの1.7%がキメラであることを発見しました。 我々の発見は、キメラがMinIONシーケンスデータにおいて一般的であることを支持する知識を追加するものである。 我々は、3つの個人および1つのマルチプレックスシーケンスデータにおいて、総リードの2~3%がキメラであることを確認しました。 本研究は、以下の2つの点で先行研究とは異なる。 まず、ライブラリ調製後、シーケンス中にキメラリードが形成されることを直接的に証明した。さらに、これらのキメラは、上述のようにマルチプレックスMinIONシーケンスにおけるクロスサンプル汚染に関連する。 一方、我々の実験のセットアップでは、ライブラリー調製時、特に標準的なマルチプレックスシーケンスプロトコルのアダプターライゲーションステップ中に形成されるキメラの可能性を特定することに限界がある。 また、ライゲーションシーケンスキット1D(SQK-LSK108)やネイティブバーコーディングキット1D(EXP-93 NBD103)など、新しい代表的なONTシーケンスキットを用いたため、MinIONシーケンサーの現状を反映した知見であった。 ナノポアシーケンス技術は急速に発展しており、あらゆる側面で改良が進んでいます。 例えば、DNAライゲーションシーケンスキット(SQK-LSK109)、ダイレクトRNAシーケンスキット(SQK-RNA001)のリリース、AlbacoreやGuppyベースコーラに搭載されているベースコールアルゴリズムのアップグレードなどがあります。 これらの変更は、Nanoporeシーケンスデータ中のキメラやマルチプレックスシーケンス時のクロスバーコードコンタミネーションに影響を与えています。 本研究の限界は実験数が少ないことであり、異なる実験セットアップを用いた追加研究は、Nanoporeマルチプレックスシーケンスデータに関する我々の理解を深めることになるであろう。 また、クロスバーコード汚染に対する潜在的な要因の寄与を調査することは重要であり、マルチプレックスシーケンスデータを解析するためのベストプラクティスに光を当てることになる。
著者による貢献
SP、KL、SL、YXがMinIONシーケンスを実施した。 YXはデータを解析した。
資金提供
本研究は、NIHR Oxford Biomedical Research Centreの支援により行われた。
利益相反声明
著者らは、潜在的な利益相反と解釈される商業的または金銭的関係がない状態で研究が行われたことを宣言します。
謝辞
フェレットの鼻腔吸引液を提供していただいたAnthony Marriott博士(Public Health England)に感謝します。