- Introducere
- Materiale și metode
- Pregătirea probelor
- Prepararea bibliotecii MinION și secvențierea
- Analiză genomică
- Rezultate
- Date de secvențiere MinION și asamblare a genomurilor virale
- Extensiunea și sursa contaminării încrucișate a probelor
- Discuție
- Contribuții ale autorilor
- Finanțare
- Declarație privind conflictul de interese
- Recunoștințe
- Material suplimentar
- Note de subsol
- Note de subsol
Introducere
Secvențierea metageneromică are potențialul de a permite identificarea nepărtinitoare a agenților patogeni dintr-o probă clinică. Aceasta promite să servească drept un test unic și universal pentru diagnosticarea bolilor infecțioase direct din probe, fără a fi nevoie de cunoștințe a priori (Bibby, 2013; Miller et al., 2013; Schlaberg et al., 2017). Pe lângă identificarea speciilor de agenți patogeni, datele de secvențe metagenomice ample și profunde ar putea furniza informații relevante pentru determinarea tratamentului și a prognosticului, detectarea focarelor și urmărirea epidemiologiei infecțiilor (Greninger et al., 2010; Yang et al., 2011; Qin et al., 2012; Loman et al., 2013). Platformele de secvențiere de generație următoare (NGS) pot produce un volum masiv de date la un cost modest; cu toate acestea, aplicarea sa în diagnosticul clinic și în sănătatea publică a fost limitată de complexitate, lentoare și investiții de capital.
MinION este un secvențiator genomic în timp real, de mărimea unei palme, în timp real, cu o singură moleculă, dezvoltat de Oxford Nanopore Technologies (ONT). Dimensiunea compactă și natura în timp real a MinION ar putea facilita aplicarea secvențierii metagenomice în cadrul testelor la locul de îngrijire pentru bolile infecțioase, așa cum au demonstrat mai multe studii de validare a conceptului, inclusiv identificarea Chikungunya (CHIKV), Ebola (EBOV) și a virusului hepatitei C (HCV) din probele de sânge clinic uman fără îmbogățire a țintei (Greninger et al., 2015) și detectarea agenților patogeni bacterieni din probele de urină (Schmidt et al., 2016) și din probele respiratorii, fără a fi necesară o cultură prealabilă (Pendleton et al., 2017).
Durata de date a MinION a crescut foarte mult de la lansarea sa în 2015, fiecare celulă de flux consumabilă generând acum până la 10-20 Gb de date de secvențe de ADN. Acest lucru le permite utilizatorilor să utilizeze mai eficient celula de flux (și să reducă costurile) prin multiplexarea mai multor probe într-o singură sesiune de secvențiere. ONT a dezvoltat seturi de coduri de bare fără PCR care permit multiplexarea a până la 12 probe.
Detecția virusului gripal A în mai multe probe respiratorii ar putea fi una dintre utilizările diagnostice ale unui test de secvențiere MinION multiplexat. Cu toate acestea, atunci când se secvențiază direct din probe cu o gamă largă potențială de titluri virale, este important să se țină seama de potențialul de contaminare încrucișată a probelor, atât în timpul pregătirii bibliotecilor, cât și în etapa de demultiplexare bioinformatică a codurilor de bare după secvențiere. Aici, prezentăm un set unic de date de secvențiere MinION și rezultatele investigației privind amploarea și sursa contaminării încrucișate a codurilor de bare în secvențierea multiplex.
Materiale și metode
Am folosit o probă de spălare nazală de dihor infectată cu virusul gripal A ca exemplu și, de asemenea, am îmbogățit două alicote de probe negative de spălare nazală de dihor neinfectate (stocuri preexistente neutilizate dintr-un studiu fără legătură) cu virusurile dengue și chikungunya separat. Niciunul dintre aceste virusuri nu este relevant pentru diagnosticul clinic în probele respiratorii, dar acționează aici ca marker clar și distinct pentru evaluarea contaminării încrucișate a probelor. Bibliotecile de secvențiere pentru fiecare probă au fost pregătite în paralel, împreună cu un martor negativ de spălare nazală, au primit coduri de bare și au fost secvențiate individual. Apoi am pus în comun o parte alicotă din bibliotecile de secvențiere și am efectuat secvențierea MinION multiplex. Lecturile din cele patru serii individuale (denumite „CHIKV”, „DENV”, „FLU-A” și „Negativ”) și din seria multiplex (denumită „Multiplexed”) au fost apoi analizate pentru a investiga amploarea și sursa contaminării încrucișate a probelor.
Pregătirea probelor
Licența proiectului a fost revizuită de către AWERB (Animal Welfare and Ethics Review Board) local și a fost ulterior acordată de către Home Office. ARN-ul a fost extras, cu ajutorul kitului de ARN viral QIAamp (Qiagen), în conformitate cu instrucțiunile producătorului, din spălături nazale de dihor care conțineau virusul gripei A (H1N1) (A/California/04/2009) și dintr-un grup de probe de spălături nazale negative. În alicote de extract din probele negative s-a adăugat fie ARN viral dengue (DENV) (tulpina TC861HA, GenBank: MF576311), fie ARN viral CHIKV (tulpina S27, GenBank: MF580946.1) din Colecția națională de viruși patogeni1. Probele au fost tratate cu DNază folosind TURBO DNază (Thermo Fisher Scientific, Waltham, MA, Statele Unite) și purificate folosind kitul RNA Clean & ConcentratorTM-5 (Zymo Research). ADNc a fost preparat și amplificat folosind metode de amplificare Sequence-Independent-Single-Primer-Amplification (Greninger et al., 2015) modificate așa cum au fost descrise anterior (Atkinson et al., 2016). ADNc amplificat a fost cuantificat cu ajutorul kitului Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA, Statele Unite), iar 1 μg a fost utilizat ca intrare pentru fiecare preparare de bibliotecă MinION, cu excepția controlului negativ, unde a fost utilizată întreaga probă (32 ng).
Prepararea bibliotecii MinION și secvențierea
Ligation Sequencing Kit 1D (SQK-LSK108) și Native Barcoding Kit 1D (EXP-NBD103) au fost utilizate în conformitate cu protocoalele standard ONT, cu excepția faptului că doar un singur cod de bare a fost inclus în fiecare dintre cele patru preparate de bibliotecă. Fiecare bibliotecă a fost rulată pe o celulă de flux individuală, iar o a cincea bibliotecă grupată a fost realizată prin combinarea celor patru biblioteci cu coduri de bare individuale. Bibliotecile au fost secvențiate pe celule de flux R9.4. Proiectul de studiu este prezentat în figura 1.
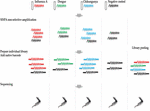
FIGURA 1. Prezentare generală a designului studiului. ARN-ul a fost extras din patru probe, inclusiv o probă de spălare nazală a dihorului infectată cu virusul gripal A, două probe negative de spălare nazală a dihorului îmbibate cu virusuri dengue și chikungunya și un martor negativ de spălare nazală a dihorului. ADNc a fost preparat și amplificat utilizând o metodă de amplificare cu un singur polimer independent de secvență (Sequence-Independent-Single-Primer-Amplification). Bibliotecile de secvențiere pentru fiecare probă au fost pregătite în paralel, cu cod de bare și secvențiate pe celule de flux individuale. Secvențierea multiplex a fost, de asemenea, realizată prin punerea în comun a celor patru biblioteci individuale. Citirile din cele patru serii individuale și din seria multiplex au fost analizate pentru a evalua amploarea și sursa de contaminare cu coduri de bare încrucișate în secvențierea multiplex.
Analiză genomică
Citirile au fost apelate de bază folosind Albacore v2.1.7 (ONT) cu demultiplexare a codurilor de bare. Lecturile din fiecare rulare de secvențiere au fost mapate la secvențele genomice ale fiecărui virus utilizând Minimap2 (Li, 2018). Numărul de citiri cartografiate la referință a fost numărat cu ajutorul Pysam2. Asamblarea de novo a fost realizată utilizând Canu v1.7 (Koren et al., 2017), iar proiectul de genom rezultat a fost șlefuit utilizând Nanopolish (Mongan et al., 2015) cu datele la nivel de semnal.
Pentru a permite demultiplexarea strictă a codurilor de bare a datelor de secvențiere multiplexă MinION, am efectuat două runde de analize utilizând Porechop (v0.2.23). Prezența secvenței adaptoare în mijlocul unei lecturi este o semnătură a chimera. Am utilizat Porechop pentru a examina fiecare citire, iar cele care au regiunea din mijloc care împărtășește >75% identitate cu secvența adaptorului au fost identificate ca lecturi chimerice. În Porechop, am setat opțiunea „-middle_threshold” și am ales un prag de 75. În a doua rundă, am utilizat Porechop pentru a căuta secvența codului de bare atât la începutul, cât și la sfârșitul unei lecturi; lecturile au fost atribuite numai dacă același cod de bare a fost găsit la cele două capete. Am setat opțiunea „-require_two_barcodes” în Porechop și am stabilit pragul pentru scorul codului de bare la 70. Pentru a găsi semnătura potențială a citirilor chimerice, am examinat semnalele de curent de citire stocate în fișierul FAST5 de către secvențiatorul MinION. Semnalele de curent au fost extrase cu ajutorul ONT fast5 API4 și reprezentate grafic cu ajutorul ggplot2 implementat în R5 pentru o comparație a citirilor chimerice și nechimerice.
Rezultate
Durata fiecărui ciclu de secvențiere MinION a variat din cauza diferențelor de timp de execuție. Un număr maxim de ∼2,4 M de citiri a fost atins de seria de secvențiere multiplexată și de seria individuală CHIKV, din cauza timpilor de execuție mai lungi (tabelul suplimentar S1). Lecturile provenite de la virusul îmbogățit au reprezentat 96 % din datele din ciclurile individuale de secvențiere CHIKV și DENV, și 78 % pentru proba FLU-A (tabelul 1). Procentul de citiri virale din cadrul fiecărei probe cu cod de bare în datele de secvențiere Multiplexed este apropiat de cel din datele probelor rulate individual (tabelul 2). Fiecare genom viral a avut o profunzime de acoperire medie ultra-înaltă (>8.000) în datele de secvențiere individuală și multiplexată, iar asamblarea de novo a fost capabilă să recupereze genomuri aproape complete pentru toate cele trei virusuri cu 99.9 % identități în comparație cu referința GenBank.

TABLĂ 1. Rezumatul rezultatelor cartografierii și asamblării de novo pentru datele provenite din secvențierea MinION a bibliotecilor individuale.

TABLĂ 2. Rezumatul rezultatelor cartografierii și asamblării de novo pentru datele provenite din secvențierea multiplexată MinION.
Extensiunea și sursa contaminării încrucișate a probelor
Care probă a fost codată cu coduri de bare și secvențiată atât individual, cât și multiplexată, ceea ce ne-a permis să examinăm performanța demultiplexării codurilor de bare ale tonului alb. În datele eșantioanelor secvențiate individual ne-am aștepta să fie prezent doar un singur cod de bare nativ. Pentru seriile de secvențiere individuală a CHIKV (cod de bare NB01), DENV (NB09) și FLU-A (NB10), am constatat că 86, 109 și, respectiv, 17 citiri au fost atribuite unor intervale de coduri de bare care nu se așteptau să fie prezente în bibliotecă (reprezentând 0,0036, 0,0129 și 0,001% din totalul citirilor). În datele de secvențiere multiplexă, 41 de citiri (0,0016 %) au fost atribuite unor coduri de bare care nu au fost incluse în experimente (adică un alt cod de bare decât NB01, NB05, NB09 sau NB10). Le-am definit ca fiind lecturi atribuite greșit (Figura 2A).
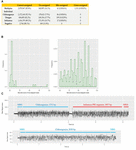
FIGURA 2. (A) rezumatul numărului și procentului de citiri atribuite corect, neatribuite, atribuite greșit și atribuite încrucișat în fiecare ciclu de secvențiere. Neatribuite se referă la citirile care nu pot fi atribuite de Albacore la niciun binișor din cauza unui scor al codului de bare mai mic de 60, atribuite greșit se referă la citirile care au fost atribuite la binișoare cu coduri de bare care nu au fost incluse în acest experiment, iar atribuite încrucișat se referă la citirile care au fost atribuite la binișoare cu coduri de bare incorecte; (B) distribuția scorurilor codurilor de bare raportate de Albacore pentru citirile atribuite greșit și citirile atribuite încrucișat în datele de secvențiere multiplex; (C) comparație a semnalului brut al unei citiri chimerice și al unei citiri atribuite corect. Semnalul citirii chimerice posedă un semnal de stagnare și un semnal de vârf imens în mijlocul citirii.
Pentru a examina posibila contaminare de laborator în pregătirea bibliotecilor de secvențiere, am cartografiat toate citirile din fiecare rulare individuală în raport cu secvențele genomice ale celor trei virusuri. Nu s-a găsit nicio citire care să provină dintr-un genom preparat într-o altă bibliotecă, ceea ce sugerează că nu există contaminare in vitro. Biblioteca de secvențiere multiplex a fost pregătită prin combinarea bibliotecilor individuale, necontaminate, după ligarea codului de bare și a adaptorului. Cu toate acestea, rezultatele cartografierii arată că 1 311 (0,0543%) lecturi au fost cartografiate la genomul țintă incorect, ceea ce implică faptul că au fost atribuite încrucișat la binișoarele de cod de bare greșite (denumite ulterior „lecturi atribuite încrucișat”), în ciuda faptului că biblioteca de secvențiere multiplexată a fost pusă în comun cu bibliotecile individuale nu a prezentat deloc lecturi atribuite încrucișat. Am emis ipoteza că lecturile atribuite greșit și cele atribuite încrucișat se datorau unui scor scăzut al codului de bare și am investigat scorurile codului de bare ale acestor lecturi. Majoritatea citirilor atribuite greșit au avut un scor al codului de bare <70, cu toate acestea, citirile atribuite încrucișat au avut scoruri mai diverse, variind de la 60 la aproape 100 (Figura 2B). Acest rezultat a sugerat că lecturile atribuite greșit și cele atribuite încrucișat provin din surse diferite. Am efectuat o analiză a citirilor atribuite încrucișate cu o mică bază de date care cuprinde secvențele genomice ale celor trei virusuri incluse în acest studiu și am demonstrat că 1074/1311 (82 %) dintre aceste citiri au putut fi aliniate încrucișat la mai mult de un genom viral (1 047 de citiri) sau aliniate încrucișat la regiuni distincte din cadrul aceluiași genom (27 de citiri), ceea ce sugerează că acestea sunt chimere. Pentru a confirma această observație, am investigat semnalele de curent brut ale câtorva lecturi aliniate încrucișate în comparație cu cele ale lecturilor corect alocate (Figura 2C). Semnalele de curent ale unei lecturi atribuite corect includ de obicei: (i) un semnal de por deschis de curent ridicat care reprezintă timpul în care porul de secvențiere trece de la un adaptor la altul, (ii) un semnal de stagnare, care se referă la perioada de timp în care o secvență de ADN se află în por, dar încă nu s-a deplasat, și (iii) urma semnalului de secvențiere a ADN-ului. În schimb, o citire chimeră posedă un semnal de blocaj și un semnal de vârf uriaș în mijlocul citirii. Lecturile chimerice pot poseda două secvențe de coduri de bare diferite la început și la sfârșit, ceea ce confundă atribuirea unui bin de coduri de bare. Luate împreună, aceste date demonstrează două categorii de erori care contribuie la contaminarea încrucișată a probelor în setul nostru de date: (i) citirile chimerice (reprezintă ∼80% din toate citirile cu atribuire încrucișată); (ii) citirile cu un scor scăzut al codului de bare. Pentru a îmbunătăți calitatea setului nostru final de date, am explorat impactul diferitelor abordări de demultiplexare a codurilor de bare pentru a elimina citirile cu atribuire încrucișată (tabelul 3). Filtrarea citirilor care posedă un adaptor intern poate elimina 90 % din citirile cu atribuire încrucișată și a pierdut 24 % din totalul citirilor. Am încercat, de asemenea, o schemă de filtrare mai strictă care necesita două coduri de bare (câte unul la începutul și la sfârșitul citirii) pentru a face o atribuire. Această abordare a eliminat toate citirile atribuite încrucișat, cu excepția a două, dar a pierdut 56% din totalul citirilor.

TABEL 3. Eliminarea citirilor cu atribuire încrucișată și pierderea datelor totale de secvențiere prin două abordări de filtrare folosind Porechop.
De asemenea, investigăm amploarea potențialelor citiri chimerice în datele de secvențiere. Pentru ciclurile individuale de secvențiere CHIKV, DENV și FLU-A, rezultatele cartografierii arată că 2,3, 3,0 și, respectiv, 2,7 % din citirile cartografiate posedă o aliniere suplimentară și s-au aliniat de cel puțin două ori la același genom (tabelul 4). Luăm în considerare atât citirile clasificate cu cod de bare, cât și cele neclasificate în datele de secvențiere multiplex. Rezultatele arată că 2,0% din citirile cartografiate posedă o aliniere suplimentară și s-au aliniat de cel puțin două ori la același genom, în timp ce 0,052% din totalul citirilor au fost aliniate la cel puțin două genomuri distincte.

TABELĂ 4. Rezumatul numărului și procentului de lecturi nechimerice, autochimerice și crosschimerice în fiecare ciclu de secvențiere.
Discuție
Obiectivul final al cercetării noastre este de a dezvolta un test de diagnostic bazat pe secvențierea metagenomică nanoporoasă care să permită testarea punctuală a bolilor infecțioase. Secvențierea multiplex oferă oportunitatea de a îmbunătăți scalabilitatea și de a reduce costurile, cu toate acestea, contaminarea încrucișată a probelor poate duce la erori în date și la o interpretare falsă a rezultatelor.
În acest experiment, am grupat biblioteci curate și am efectuat secvențierea multiplex MinION pentru a investiga amploarea și sursa contaminării cu coduri de bare încrucișate. Am identificat că 0,056 % din totalul citirilor au fost atribuite încrucișat la bini de coduri de bare incorecte, ceea ce este comparabil cu cele raportate pentru platformele de secvențiere Illumina din diferite studii (între 0,06 și 0,25 %) (Nelson et al., 2014; D’Amore et al., 2016; Wright și Vetsigian, 2016). Rezultatele noastre au arătat că citirile chimerice sunt sursa predominantă a erorilor de atribuire a codurilor de bare încrucișate. Lecturile chimerice cu atribuire încrucișată din acest set de date ar fi putut fi formate doar în timpul secvențierii, mai degrabă decât în timpul pregătirii bibliotecilor, deoarece acestea au fost complet absente în datele de secvențiere ale bibliotecilor individuale, iar singura etapă de procesare ulterioară a fost amestecarea bibliotecilor de secvențiere finale înainte de încărcare. Formulăm ipoteza că algoritmul actual implementat în Albacore nu poate recunoaște disocierea scurtă dintre secvențele de ADN care trec concomitent prin nanopor, concatenând astfel mai multe secvențe în același fișier Fast5.
Lecturi chimerice au fost observate în datele de secvențiere MinION înainte în White et al. (2017). Prin analizarea datelor de secvențiere MinION a trei ampliconi diferiți de interferon, autorii au constatat că 1,7 % din citirile cartografiate erau chimera. Constatările noastre se adaugă la cunoștințele care susțin că chimera sunt frecvente în datele de secvențiere MinION. Am identificat între 2 și 3% din totalul citirilor în trei date de secvențiere individuală și una de secvențiere multiplex sunt chimera. Studiul nostru diferă de lucrările anterioare în următoarele două aspecte. În primul rând, furnizăm dovezi directe că citirile chimerice se pot forma după pregătirea bibliotecii și în timpul secvențierii; de asemenea, am legat aceste chimera de contaminarea încrucișată a eșantioanelor în secvențierea MinION multiplex, așa cum am discutat mai sus. Pe de altă parte, configurația experimentului nostru are o limitare în identificarea potențialelor chimera formate în timpul pregătirii bibliotecii, în special în timpul etapei de ligare a adaptorului în protocolul standard de secvențiere multiplex. În al doilea rând, constatările noastre reflectă stadiul actual al secvențierea MinION, deoarece am utilizat cele mai noi și mai reprezentative kituri de secvențiere ONT, inclusiv kitul de secvențiere prin ligaturare 1D (SQK-LSK108) și kitul de barcodare nativă 1D (EXP-93 NBD103). Tehnologia de secvențiere cu nanopori este în curs de dezvoltare rapidă și se produc îmbunătățiri în toate aspectele. De exemplu, au fost lansate un kit mai nou de secvențiere prin ligatura ADN (SQK-LSK109) și un kit de secvențiere directă a ARN-ului (SQK-RNA001); algoritmul de basecalling implementat în Albacore și Guppy basecaller a fost actualizat. Toate aceste modificări au efect asupra gradului de chimeră în datele de secvențiere Nanopore și asupra contaminării cross-barcode în timpul secvențierii multiplex. Limitarea acestui studiu a fost reprezentată de numărul mic de experimente; lucrări suplimentare care utilizează diferite configurații de experimente ar contribui la înțelegerea datelor de secvențiere multiplexă Nanopore. În plus, este important să se investigheze contribuțiile factorilor potențiali la contaminarea cu coduri de bare încrucișate, ceea ce ar pune în lumină cele mai bune practici de analiză a datelor de secvențiere multiplex.
În concluzie, studiul nostru a demonstrat că citirile chimerice sunt sursa predominantă a erorilor de atribuire a codurilor de bare încrucișate în secvențierea multiplexă MinION. Acesta evidențiază necesitatea unei filtrări atente a datelor de secvențiere MinION multiplex înainte de analiza în aval, precum și compromisul dintre sensibilitate și specificitate care se aplică metodelor de demultiplexare a codurilor de bare.
Contribuții ale autorilor
SP, KL, SL și YX au efectuat secvențierea MinION. YX a analizat datele. Toți autorii au conceput studiul, au participat la interpretarea rezultatelor și la redactarea manuscrisului și au citit și aprobat versiunea finală a acestui manuscris.
Finanțare
Această lucrare a fost susținută de NIHR Oxford Biomedical Research Centre.
Declarație privind conflictul de interese
Autorii declară că cercetarea a fost efectuată în absența oricăror relații comerciale sau financiare care ar putea fi interpretate ca un potențial conflict de interese.
Recunoștințe
Am dori să mulțumim Dr. Anthony Marriott (Public Health England) pentru furnizarea de aspirate nazale de dihor.