Úvod
Metagenomické sekvenování má potenciál umožnit objektivní identifikaci patogenů z klinického vzorku. Je příslibem, že bude sloužit jako jediný a univerzální test pro diagnostiku infekčních onemocnění přímo ze vzorků bez nutnosti apriorních znalostí (Bibby, 2013; Miller et al., 2013; Schlaberg et al., 2017). Kromě identifikace druhů patogenů by rozsáhlá a hluboká metagenomická sekvenční data mohla poskytnout informace důležité pro stanovení léčby a prognózy, odhalení ohnisek a sledování epidemiologie infekcí (Greninger et al., 2010; Yang et al., 2011; Qin et al., 2012; Loman et al., 2013). Platformy pro sekvenování nové generace (NGS) mohou produkovat obrovskou kapacitu dat při nízkých nákladech, nicméně jejich použití v klinické diagnostice a veřejném zdravotnictví je omezeno složitostí, pomalostí a kapitálovými investicemi.
MinION je sekvenátor genomu o velikosti dlaně, pracující v reálném čase s jednou molekulou, který vyvinula společnost Oxford Nanopore Technologies (ONT). Kompaktní rozměry a povaha přístroje MinION v reálném čase by mohly usnadnit použití metagenomického sekvenování při testování infekčních onemocnění v místě péče, jak prokázalo několik studií proof-of-concept, včetně identifikace virů Chikungunya (CHIKV), Ebola (EBOV) a viru hepatitidy C (HCV) z klinických vzorků lidské krve bez obohacování cílů (Greninger et al..), 2015) a detekce bakteriálních patogenů ze vzorků moči (Schmidt et al., 2016) a respiračních vzorků bez nutnosti předchozí kultivace (Pendleton et al., 2017).
Propustnost dat systému MinION se od jeho uvedení na trh v roce 2015 výrazně zvýšila, přičemž každá spotřební průtoková buňka nyní generuje až 10-20 Gb sekvenčních dat DNA. To uživatelům umožňuje efektivnější využití průtokové buňky (a snížení nákladů) multiplexováním několika vzorků v jednom sekvenačním běhu. Společnost ONT vyvinula sady čárových kódů bez PCR, které umožňují multiplexovat až 12 vzorků.
Detekce viru chřipky A ve více respiračních vzorcích by mohla být jedním z diagnostických využití multiplexovaného sekvenovacího testu MinION. Při sekvenování přímo ze vzorků s potenciálně širokým rozsahem virových titrů je však důležité mít na paměti možnost křížové kontaminace vzorků, a to jak během přípravy knihovny, tak ve fázi bioinformatické demultiplexace čárových kódů po sekvenování. Zde představujeme jedinečný soubor dat sekvenování MinION a výsledky zkoumání rozsahu a zdroje křížové kontaminace čárovým kódem při multiplexním sekvenování.
Materiál a metody
Jako vzorový vzorek jsme použili vzorek výplachu nosu fretky infikované virem chřipky A a také jsme zvlášť spikovali dva alikvoty negativních vzorků výplachu nosu neinfikované fretky (již existující nepoužité zásoby z nesouvisející studie) viry dengue a chikungunya. Ani jeden z těchto virů není relevantní pro klinickou diagnostiku v respiračních vzorcích, ale působí zde jako jasné, odlišné markery pro posouzení křížové kontaminace vzorků. Sekvenační knihovny pro každý vzorek byly připraveny paralelně spolu s negativní kontrolou z nosního výplachu, opatřeny čárovým kódem a sekvenovány jednotlivě. Poté jsme spojili alikvotní část sekvenačních knihoven a provedli multiplexní sekvenování MinION. Čtení ze čtyř individuálních běhů (označovaných jako „CHIKV“, „DENV“, „FLU-A“ a „Negativní“) a multiplexního běhu (označovaného jako „Multiplexed“) byla poté analyzována za účelem zkoumání rozsahu a zdroje křížové kontaminace vzorků.
Příprava vzorků
Licence projektu byla posouzena místní komisí AWERB (Animal Welfare and Ethics Review Board) a následně byla udělena ministerstvem vnitra. RNA byla extrahována pomocí soupravy QIAamp pro virovou RNA (Qiagen) podle pokynů výrobce z nosních výplachů fretek obsahujících virus chřipky A (H1N1) (A/California/04/2009) a ze souboru negativních vzorků nosních výplachů. Do alikvotů extraktu negativních vzorků byla přidána virová RNA viru dengue (DENV) (kmen TC861HA, GenBank: MF576311) nebo CHIKV (kmen S27, GenBank: MF580946.1) z Národní sbírky patogenních virů1. Vzorky byly ošetřeny DNázou pomocí TURBO DNázy (Thermo Fisher Scientific, Waltham, MA, Spojené státy) a přečištěny pomocí soupravy RNA Clean & ConcentratorTM-5 (Zymo Research). cDNA byla připravena a amplifikována pomocí metody Sequence-Independent-Single-Primer-Amplification (Greninger et al., 2015) upravené podle předchozího popisu (Atkinson et al., 2016). Amplifikovaná cDNA byla kvantifikována pomocí sady Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA, Spojené státy) a jako vstup pro každou přípravu knihovny MinION bylo použito 1 μg, s výjimkou negativní kontroly, kde byl použit celý vzorek (32 ng).
Příprava knihoven MinION a sekvenování
Sada pro sekvenování ligací 1D (SQK-LSK108) a sada pro nativní čárové kódy 1D (EXP-NBD103) byly použity podle standardních protokolů ONT s tou výjimkou, že v každé ze čtyř příprav knihoven byl zahrnut pouze jeden čárový kód. Každá knihovna byla spuštěna na samostatné průtokové buňce a pátá sdružená knihovna byla vytvořena spojením čtyř individuálně čárově kódovaných knihoven. Knihovny byly sekvenovány na průtokových buňkách R9.4. Plán studie je znázorněn na obrázku 1.
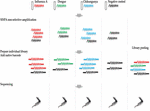
Obr. 1. Přehled designu studie. RNA byla extrahována ze čtyř vzorků, včetně vzorku nosního výplachu fretky infikované virem chřipky A, dvou negativních vzorků nosního výplachu fretky s příměsí virů dengue a chikungunya a negativní kontroly nosního výplachu fretky. cDNA byla připravena a amplifikována metodou Sequence-Independent-Single-Primer-Amplification. Sekvenační knihovny pro každý vzorek byly připraveny paralelně, opatřeny čárovým kódem a sekvenovány na jednotlivých průtokových buňkách. Multiplexní sekvenování bylo rovněž provedeno spojením čtyř jednotlivých knihoven. Čtení ze čtyř jednotlivých běhů a multiplexního běhu byla analyzována za účelem posouzení rozsahu a zdroje křížové kontaminace čárovým kódem při multiplexním sekvenování.
Genomická analýza
Čtení byla bazálně vyvolána pomocí programu Albacore v2.1.7 (ONT) s demultiplexací čárového kódu. Čtení z každého běhu sekvenování byla mapována na genomové sekvence jednotlivých virů pomocí programu Minimap2 (Li, 2018). Počet čtení mapovaných k referenčním byl spočítán pomocí programu Pysam2. Sestavení de novo bylo provedeno pomocí Canu v1.7 (Koren et al., 2017) a výsledný draft genomu byl vyleštěn pomocí Nanopolish (Mongan et al., 2015) s daty na úrovni signálu.
Abychom umožnili přísnou demultiplexaci čárových kódů dat multiplexního sekvenování MinION, provedli jsme dvě kola analýz pomocí Porechop (v0.2.23). Přítomnost sekvence adaptéru uprostřed čtení je znakem chiméry. Pomocí programu Porechop jsme prozkoumali každý čtení a ta, která mají střední oblast sdílející >75% identitu se sekvencí adaptéru, byla identifikována jako chimérická čtení. V programu Porechop jsme nastavili možnost „-middle_threshold“ a zvolili jsme prahovou hodnotu 75. Ve druhém kole jsme pomocí programu Porechop vyhledali sekvenci čárového kódu na začátku i na konci čtení; čtení byla přiřazena pouze tehdy, pokud byl na obou koncích nalezen stejný čárový kód. V programu Porechop jsme nastavili volbu „-require_two_barcodes“ a stanovili jsme práh pro skóre čárového kódu na 70. Abychom našli potenciální podpis chimérických čtení, zkoumali jsme signály proudu čtení uložené v souboru FAST5 sekvenátorem MinION. Aktuální signály byly extrahovány pomocí ONT fast5 API4 a vykresleny pomocí ggplot2 implementovaného v R5 pro srovnání chimérických a nechimérických čtení.
Výsledky
Data sekvenování MinION a sestavení virových genomů
Propustnost jednotlivých běhů sekvenování MinION se lišila kvůli rozdílům v době běhu. Maximálního počtu ∼2,4 M čtení bylo dosaženo při multiplexním sekvenačním běhu a individuálním běhu CHIKV, a to v důsledku delší doby běhu (doplňková tabulka S1). Čtení z nasypaného viru tvořila 96 % dat v jednotlivých bězích sekvenování CHIKV a DENV a 78 % u vzorku FLU-A (tabulka 1). Procento čtení z viru v rámci každého vzorku s čárovým kódem v datech multiplexního sekvenování se blíží procentu v datech z jednotlivě spuštěných vzorků (tabulka 2). Každý virový genom měl ultra vysokou (>8 000) střední hloubku pokrytí v datech individuálního a multiplexního sekvenování a sestavením de novo se podařilo získat téměř kompletní genomy pro všechny tři viry s 99.

Tabulka 1. Shrnutí výsledků mapování a de novo sestavení pro data ze sekvenování MinION jednotlivých knihoven.

TABULKA 2. Výsledky mapování a de novo sestavení pro data ze sekvenování MinION jednotlivých knihoven. Shrnutí výsledků mapování a de novo sestavení pro data z multiplexního sekvenování MinION
Rozsah a zdroj kontaminace křížových vzorků
Každý vzorek byl opatřen čárovým kódem a sekvenován jak individuálně, tak multiplexně, což nám umožnilo prozkoumat výkonnost demultiplexování čárového kódu alba. V datech individuálně sekvenovaných vzorků bychom očekávali přítomnost pouze jednoho nativního čárového kódu. U individuálního sekvenování CHIKV (čárový kód NB01), DENV (NB09) a FLU-A (NB10) jsme zjistili, že 86, 109, resp. 17 čtení bylo přiřazeno ke košům čárových kódů, u nichž se neočekávala přítomnost v knihovně (což představuje 0,0036, 0,0129 a 0,001 % celkových čtení). V datech z multiplexního sekvenování bylo 41 čtení (0,0016 %) přiřazeno čárovým kódům, které nebyly zahrnuty do experimentů (tj. jiný čárový kód než NB01, NB05, NB09 nebo NB10). Tyto čtení jsme definovali jako chybně přiřazená čtení (Obrázek 2A).
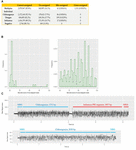
Obrázek 2. (A) Přehled počtu a procenta správně přiřazených, nezařazených, chybně přiřazených a křížově přiřazených čtení v každém běhu sekvenování. Nezařazené označuje čtení, která společnost Albacore nemůže přiřadit k žádnému binu kvůli skóre čárového kódu nižšímu než 60, chybně přiřazené označuje čtení, která byla přiřazena k binům čárového kódu nezahrnutým do tohoto experimentu, a křížově přiřazené označuje čtení, která byla přiřazena k nesprávným binům čárového kódu; (B) distribuce skóre čárového kódu hlášeného společností Albacore pro chybně přiřazená čtení a křížově přiřazená čtení v datech multiplexního sekvenování; (C) srovnání surového signálu chimérického a správně přiřazeného čtení. Signál chimérického čtení disponuje signálem stall a obrovským signálem spike uprostřed čtení.
Pro zkoumání potenciální laboratorní kontaminace při přípravě sekvenační knihovny jsme mapovali všechna čtení z každého jednotlivého běhu proti genomovým sekvencím všech tří virů. Nebylo zjištěno žádné čtení, které by pocházelo z genomu připraveného v jiné knihovně, což naznačuje, že nedošlo ke kontaminaci in vitro. Multiplexní sekvenační knihovna byla připravena spojením jednotlivých nekontaminovaných knihoven po ligaci čárového kódu i adaptéru. Výsledky mapování však ukázaly, že 1 311 (0,0543 %) čtení bylo namapováno na nesprávný cílový genom, což znamená, že byla zkříženě přiřazena do nesprávných binů čárového kódu (později označováno jako „zkříženě přiřazená čtení“), přestože multiplexní sekvenační knihovna byla spojena s jednotlivými knihovnami nevykazovala vůbec žádná zkříženě přiřazená čtení. Předpokládali jsme, že chybně přiřazené a křížově přiřazené čtení byly způsobeny nízkým skóre čárového kódu, a zkoumali jsme skóre čárového kódu těchto čtení. Většina chybně přiřazených čtení měla skóre čárového kódu <70, avšak křížově přiřazená čtení měla různorodější skóre od 60 do téměř 100 (obrázek 2B). Tento výsledek naznačuje, že chybně přiřazené a křížově přiřazené čtení pocházejí z různých zdrojů. Křížově přiřazená čtení jsme porovnali s malou databází obsahující genomové sekvence tří virů zahrnutých do této studie a prokázali jsme, že 1074/1311 (82 %) těchto čtení lze křížově přiřadit k více než jednomu virovému genomu (1047 čtení) nebo je lze křížově přiřadit k různým oblastem v rámci stejného genomu (27 čtení), což naznačuje, že se jedná o chiméry. Abychom toto pozorování potvrdili, zkoumali jsme surové proudové signály několika křížově přiřazených čtení ve srovnání se signály správně přiřazených čtení (obrázek 2C). Aktuální signály správně přiřazeného čtení obvykle obsahují: (i) signál otevřeného póru s vysokým proudem, který představuje dobu, kdy se sekvenační pór mění z jednoho adaptéru na druhý, (ii) signál zdržení, který se vztahuje k době, kdy je sekvence DNA v póru, ale ještě se nepohybuje, a (iii) stopu signálu sekvenování DNA. Naproti tomu chimérické čtení má signál stall a obrovský signál spike uprostřed čtení. Chimérické čtení může mít na začátku a na konci dvě různé sekvence čárového kódu, což mate přiřazení koše čárového kódu. Celkově tyto údaje ukazují dvě kategorie chyb, které přispívají ke kontaminaci křížových vzorků v našem souboru dat: (i) chimérické čtení (tvoří ∼80 % všech křížově přiřazených čtení); (ii) čtení s nízkým skóre čárového kódu. Abychom zlepšili kvalitu našeho konečného souboru dat, zkoumali jsme vliv různých přístupů demultiplexace čárových kódů na odstranění křížově přiřazených čtení (tabulka 3). Filtrováním čtení, která mají vnitřní adaptér, lze odstranit 90 % křížově přiřazených čtení a ztratit 24 % celkových čtení. Vyzkoušeli jsme také přísnější schéma filtrování, které pro přiřazení vyžadovalo dva čárové kódy (po jednom na začátku a na konci čtení). Tento přístup odstranil všechna křížově přiřazená čtení kromě dvou, ale ztratil 56 % celkových čtení.

Tabulka 3. Odstranění křížově přiřazených čtení a ztráta celkových sekvenačních dat pomocí dvou přístupů filtrování pomocí Porechop.
Zkoumáme také rozsah potenciálních chimérických čtení v sekvenačních datech. U jednotlivých běhů sekvenování CHIKV, DENV a FLU-A výsledky mapování ukazují, že 2,3, 3,0, resp. 2,7 % mapovaných čtení má doplňkové zarovnání a je zarovnáno alespoň dvakrát ke stejnému genomu (tabulka 4). V datech z multiplexního sekvenování bereme v úvahu jak klasifikovaná, tak neklasifikovaná čtení čárového kódu. Výsledky ukazují, že 2,0 % mapovaných čtení má doplňkové zarovnání a zarovnalo se alespoň dvakrát ke stejnému genomu, zatímco 0,052 % všech čtení bylo zarovnáno alespoň ke dvěma různým genomům.

TABULKA 4. Přehled počtu a procenta nechimérických, samochimérických a křížově chimérických čtení v každém běhu sekvenování.
Diskuse
Konečným cílem našeho výzkumu je vyvinout diagnostický test založený na nanoporovém metagenomickém sekvenování, který umožní testování infekčních onemocnění v místě péče. Multiplexní sekvenování nabízí možnost zlepšit škálovatelnost a snížit náklady, nicméně křížová kontaminace vzorků může vést k chybám v datech a chybné interpretaci výsledků.
V tomto experimentu jsme spojili čisté knihovny a provedli multiplexní sekvenování MinION s cílem prozkoumat rozsah a zdroj křížové kontaminace vzorků. Zjistili jsme, že 0,056 % celkových čtení bylo přiřazeno do nesprávných binů čárových kódů, což je srovnatelné s hodnotami uváděnými pro sekvenační platformy Illumina z různých studií (mezi 0,06 a 0,25 %) (Nelson et al., 2014; D’Amore et al., 2016; Wright a Vetsigian, 2016). Naše výsledky ukázaly, že převažujícím zdrojem chyb při přiřazování křížových kódů jsou chimérická čtení. Křížově přiřazená chimérická čtení v tomto souboru dat mohla vzniknout pouze během sekvenování, nikoliv při přípravě knihoven, protože v sekvenačních datech jednotlivých knihoven zcela chyběla a jediným dalším krokem zpracování bylo smíchání finálních sekvenačních knihoven před načtením. Předpokládáme, že současný algoritmus implementovaný v systému Albacore nedokáže rozpoznat krátkou disociaci mezi sekvencemi DNA, které procházejí nanoporem současně, a tím spojit více než jednu sekvenci do jednoho souboru Fast5.
Chimerická čtení byla pozorována v datech sekvenování MinION již dříve v práci White et al. (2017). Analýzou dat sekvenování MinION tří různých interferonových amplikonů autoři zjistili, že 1,7 % mapovaných čtení bylo chimérických. Naše zjištění doplňují poznatky podporující, že chiméry jsou v datech sekvenování MinION běžné. Zjistili jsme, že 2 až 3 % celkových čtení ve třech individuálních a jednom multiplexním sekvenačním údaji jsou chiméry. Naše studie se od předchozích prací liší v následujících dvou aspektech. Zaprvé poskytujeme přímý důkaz, že chimérická čtení mohou vznikat po přípravě knihovny a během sekvenování; dále jsme tyto chiméry spojili s křížovou kontaminací vzorků při multiplexním sekvenování MinION, jak je uvedeno výše. Na druhou stranu má naše nastavení experimentu omezení při identifikaci potenciálních chimér vznikajících při přípravě knihovny, zejména během kroku ligování adaptérů ve standardním protokolu multiplexního sekvenování. Za druhé, naše zjištění odrážejí současný stav sekvenování MinION, protože jsme použili novější a nejreprezentativnější soupravu pro sekvenování ONT, včetně soupravy pro ligační sekvenování 1D (SQK-LSK108) a soupravy pro nativní čárové kódování 1D (EXP-93 NBD103). Technologie nanoporového sekvenování se rychle vyvíjí a dochází k jejímu zdokonalování ve všech aspektech. Například byla vydána novější sada pro ligované sekvenování DNA (SQK-LSK109) a sada pro přímé sekvenování RNA (SQK-RNA001); byl vylepšen algoritmus basecallingu implementovaný v Albacore a Guppy basecalleru. Všechny tyto změny mají vliv na rozsah chimér v datech sekvenování Nanopore a na kontaminaci křížovými kódy při multiplexním sekvenování. Omezením této studie byl malý počet experimentů, další práce s použitím různých nastavení experimentů by přispěla k našemu porozumění datům z multiplexního sekvenování Nanopore. Kromě toho je důležité prozkoumat podíl potenciálních faktorů na kontaminaci křížovým čárovým kódem, což by osvětlilo osvědčené postupy při analýze dat multiplexního sekvenování.
V souhrnu naše studie prokázala, že chimérická čtení jsou převažujícím zdrojem chyb při přiřazování křížových čárových kódů při multiplexním sekvenování MinION. Zdůrazňuje potřebu pečlivého filtrování dat multiplexního sekvenování MinION před následnou analýzou a kompromis mezi citlivostí a specifičností, který platí pro metody demultiplexace čárových kódů.
Příspěvky autorů
SP, KL, SL a YX prováděli sekvenování MinION. YX analyzoval data. Všichni autoři navrhli studii, podíleli se na interpretaci výsledků a psaní rukopisu a přečetli a schválili konečnou verzi tohoto rukopisu.
Financování
Tato práce byla podpořena NIHR Oxford Biomedical Research Centre.
Prohlášení o střetu zájmů
Autoři prohlašují, že výzkum byl prováděn bez jakýchkoli komerčních nebo finančních vztahů, které by mohly být chápány jako potenciální střet zájmů.
Poděkování
Rádi bychom poděkovali Dr. Anthonymu Marriottovi (Public Health England) za poskytnutí nosních aspirátů fretek.
Přídavné materiály
Poznámky
.