Sir,
Der Morbus Darier (DD) ist eine seltene erbliche Hauterkrankung, die vor allem im Jugendalter schwer zu behandeln ist. Es wurde berichtet, dass die Krankheit durch Mutationen in einem Gen verursacht wird, das auf einem einzigen Locus auf Chromosom 12q23-q24.1 kartiert ist und für eine sarko-endoplasmatische Retikulum-Kalzium-ATPase-Pumpe (SERCA2) kodiert (1). Die DD weist charakteristische klinische und histologische Befunde auf, kann jedoch manchmal fehldiagnostiziert oder übersehen werden. Wir berichten hier über einen tödlichen Fall von DD, der in einer anderen Einrichtung 11 Jahre lang als atopische Dermatitis fehldiagnostiziert worden war. Der Patient hatte wiederholt bakterielle und psychiatrische Störungen.
FALLBERICHT
Ein 25-jähriger Japaner stellte sich am 16. Oktober 2004 mit einem schmerzhaften erosiven Hautausschlag und hohem Fieber in unserem Krankenhaus vor. Er litt seit seinem 14. Lebensjahr an einem Hautausschlag, der als atopische Dermatitis diagnostiziert und behandelt worden war. Im Laufe der Jahre waren Kortikosteroidsalben ohne signifikante klinische Reaktion verwendet worden.
Bei der körperlichen Untersuchung zeigte sich ein ausgedehntes Erythem am gesamten Körper. Er hatte auch geruchsintensive erosive Läsionen an Rumpf, Gesäß und unteren Extremitäten. Unsere erste Diagnose lautete atopische Dermatitis mit schwerer Sekundärinfektion. Er wurde in unserer Abteilung aufgenommen. In Bakterienkulturen aus seinen erosiven Hautläsionen wurden Streptococcus aureus, Pseudomonas aeruginosa und Klebsiella pneumoniae nachgewiesen. Zu den abnormen Laborbefunden gehörten eine Leukozytose (13.500/mm3), erhöhte Werte des C-reaktiven Proteins (CRP) (9,4 mg/dl) und eine Hypoproteinämie (Gesamtprotein 5,8 g/dl, Albumin 2,0 g/dl). Die Behandlung mit oralem Prednisolon 30 mg/Tag sowie systemischen und topischen antibakteriellen Mitteln führte zu einem teilweisen Ansprechen. Einige Tage später traten zahlreiche warzige hyperkeratotische Papeln im Gesicht, am Rumpf und an den Gliedmaßen auf (Abb. 1). Eine sorgfältige Untersuchung ergab außerdem V-förmige Einkerbungen an den freien Rändern seiner Fingernägel und Längsstreifen in einigen Fingernägeln. Es gab keine Grübchen an der Handfläche oder Läsionen der Mundschleimhaut.
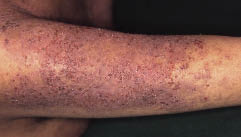
Abb. 1. Eine Woche nach der Aufnahme. Disseminierte keratotische Papeln wurden am Rumpf und an den Extremitäten der Patientin festgestellt.
Eine aus der Brust entnommene Biopsieprobe zeigte eine ausgeprägte Hyperkeratose mit Parakeratose, suprabasalen Spalten und Akantholyse in Verbindung mit Korpsronds und Körnern (Abb. 2). Die Familienanamnese ergab, dass seine Mutter und seine Schwester ebenfalls multiple keratotische Papeln aufwiesen. Daraufhin stellten wir die Diagnose einer DD. Die Behandlung mit oralem Etretinat, 40 mg/Tag, führte zu einer teilweisen Besserung. Er wurde am 25. Juli 2005 entlassen.
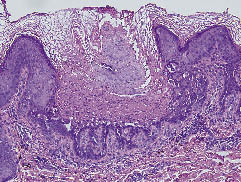
Abb. 2. Ausgeprägte Hyperkeratose mit Parakeratose, suprabasalen Spalten und Akantholyse in Verbindung mit Dyskeratose einschließlich Korpsronds und Körnern. (Hämatoxylin- und Eosinfärbung (H&E) ×200).
Sechs Wochen später wurde er wegen des Wiederauftretens von schmerzhaften erosiven Hautläsionen, begleitet von hohem Fieber und Dyspnoe, wieder aufgenommen (Abb. 3). Die Ergebnisse der Bakterienkulturen aus der erosiven Haut zeigten P. aeruginosa und andere. Die Behandlung mit intravenösem Minocyclinhydrochlorid 200 mg/Tag und die topische Anwendung einer antibakteriellen Salbe führten zu einer Besserung der Infektion. Die Papeln und das Erythem blieben unverändert, und orales Etretinat 40 mg/Tag und Cyclosporin 200 mg/Tag waren unwirksam. Etretinat wurde auf 70 mg/Tag erhöht, und Gentamicin-Salbe wurde topisch auf den gesamten Körper aufgetragen. Die Erosionen des Rumpfes besserten sich allmählich, traten dann aber wieder auf. Am 11. April 2006 versuchten wir eine Ablationsbehandlung und ein verdicktes Spalthauttransplantat an der linken unteren Extremität. Der transplantierte Bereich heilte einige Wochen lang fast vollständig ab, jedoch entwickelten sich erneut hämorrhagische und erosive Läsionen. Außerdem traten dreimal Kaposi-Varizellenausbrüche auf, begleitet von hohem Fieber und einer sich rasch ausbreitenden erosiven Haut. Jedes Mal erhielt der Patient intravenöses Aciclovir 750 mg/Tag.
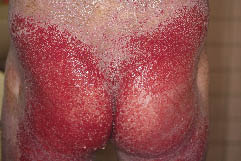
Abb. 3. Weitverbreitete Rötung des gesamten Körpers. Geruchsintensive erosive Läsionen betrafen den Rumpf, das Gesäß und die unteren Extremitäten.
Laboruntersuchungen ergaben eine anhaltende Hypoalbuminämie von 0,9 bis 1,7 mg/dl, trotz einer kalorienreichen Diät und intravenösem Albumin. Da die Darmuntersuchungen keine Auffälligkeiten ergaben, gingen wir davon aus, dass die Hypoalbuminämie durch seine stark erodierte Haut verursacht wurde.
Im Mai 2006 verschlimmerten sich die Hautläsionen so schnell, dass erneut intravenöse Antibiotika verabreicht wurden. Als Folge der Sepsis und der Dehydrierung entwickelte sich jedoch ein akutes Nierenversagen. Der Patient starb am 30. Juli 2006.
DISKUSSION
Der Schweregrad der DD ist nicht vorhersehbar, aber sie verläuft in der Regel chronisch mit Exazerbationen und Remissionen (2, 3). Exazerbationen können durch heißes Wetter, übermäßige Sonneneinstrahlung, Medikamente, Steroide oder mechanische Traumata verursacht werden. Die Häufigkeit von Schleimhaut- oder gastrointestinalen Läsionen schwankt zwischen 15 und 50 % (4).
Es gibt mehrere klinische Varianten der DD, darunter hypertrophe, lineare oder zosterförmige und vesikulo-bullöse Formen (3, 5-9). Bei der letztgenannten Form entwickeln sich Bläschen und Bullae in der exponierten Haut, die häufig durch hohe Luftfeuchtigkeit, physischen oder chirurgischen Stress und bakterielle oder virale Hautinfektionen ausgelöst werden. Lokalisierte Blasenbildung ist bei DD nicht ungewöhnlich, aber eine weit verbreitete vesikulo-bulloöse Form ist selten. Unser Fall könnte diesem Typ zugeordnet werden, und zweifellos muss eine bakterielle Sekundärinfektion, die die keratotischen Ablagerungen besiedelt, die hartnäckigen Läsionen verursacht haben.
Obwohl es einige Berichte über selektive Immundefekte bei Patienten mit Morbus Darier gibt, konnte keine einheitliche oder spezifische Anomalie nachgewiesen werden (10). Nikkels et al. (11) berichteten über einen tödlichen Fall mit schwerer HSV-Hautinfektion, gefolgt von HSV-bedingter Lungenentzündung, gastrointestinaler Beteiligung und akutem Atemnotsyndrom.
Orale Retinoide haben sich bei DD als recht wirksam erwiesen (12). Die Behandlung einer weit verbreiteten vesikulo-bullösen DD ist jedoch schwierig, da orale Retinoide die Hautbrüchigkeit erhöhen können. Andere topische Behandlungen mit 5-Fluorouracil, Tazaroten oder Calcipotriol wurden mit unterschiedlichen Ergebnissen eingesetzt. In hartnäckigen Fällen wurde auch über die systemische Verabreichung von Cyclosporin, Verhütungsmitteln und Diazepam berichtet (12, 13). Orale Steroide oder Cyclosporin reduzieren die Entzündung bei Patienten mit ekzematösen Zuständen (15), aber Papeln und Erosionen sprechen oft nicht darauf an. In einer kürzlich durchgeführten Studie konnte gezeigt werden, dass topische Aminoglykoside bei einem Patienten mit Hailey-Hailey-Krankheit eine Remission herbeiführten, indem sie die Auswirkungen der pathogenen Nonsense-Mutationen rückgängig machten (14). Wie in dieser Studie versuchten wir es mit einer topischen Gentamicin-Salbe. Die Exsudation ging zwar allmählich zurück, doch reichte dies nicht aus, um die Hautläsionen zum Abklingen zu bringen. Photodynamische Therapie, Exzision, Elektrodessikation, Dermabrasion, Abrasion mit Kohlendioxid- oder Erbium-YAG-Laser haben sich als erfolgreich erwiesen (3). Die Dermabrasion und die Spalthauttransplantation waren bei unserem Patienten, der schließlich aufgrund schwerer systemischer Komplikationen starb, nicht wirksam.
1. Sakuntabhai A, Ruiz-Perez V, Carter S, Jacobsen N, Burge S, Monk S, et al. Mutationen in ATP2A2, das für eine Ca2+-Pumpe kodiert, verursachen das Darier-Syndrom. Nat Genet 1999; 21: 271-277.
2. Burge SM, Wilkinson JD. Darier-White-Krankheit: ein Überblick über die klinischen Merkmale bei 163 Patienten. J Am Acad Dermatol 1992; 27: 40-50.
3. Sehgal VN, Srivastava G. Darier’s (Darier-White) disease/ keratosis follicularis. Int J Dermatol 2005; 44: 184-192.
4. Robaee AA, Hamadah LR, Khuroo S, Alfadley A. Extensive Darier-Krankheit mit Beteiligung der Speiseröhre. Int J Dermatol 2004; 43: 835-839.
5. Mei S, Amato L, Gallerani I, Perrella E, Caproni M, Palleschi GM, Fabbri P. Ein Fall von vesikulo-bullösem Morbus Darier in Verbindung mit einer bipolaren psychiatrischen Störung. J Dermatol 2000; 27: 673-676.
6. Telfer NR, Burge SM, Ryan TJ. Vesiculo-bullous Darier’s disease. Br J Dermatol 1990; 122: 831-834.
7. Hori Y, Tusuru N, Niimura M. Bullous Darier’s disease. Arch Dermatol 1982; 118: 278-279.
8. Colver GB, Gawkrodger DJ. Vesiculo-bullous Darier’s disease. Br J Dermatol 1992; 126: 416-417.
9. Speight EL. Vesikulo-bullöser Morbus Darier, der auf orale Prednisolon-Steroide anspricht. Br J Dermatol 1998; 139: 934-935.
10. Partrizi A, Ricci G, Neri I, Specchia F, Varotti C, Masi M. Immunological parameters in Darier’s disease. Dermatologica 1989; 178: 138-140.
11. Nikkels AF, Beauthier F, Quatresooz P, Pierard GE. Tödliche Herpes-simplex-Virusinfektion bei Morbus Darier unter Kortikostherapie. Eur J Dermatol 2005; 15: 293-297.
12. Burge S. Management of Darier’s Disease. Clin Exp Dermatol 1999; 24: 53-56.
13. Shahidullah H, Humphreys F, Beveridge GW. Darier-Krankheit: Schwere Ekzematisierung erfolgreich mit Cyclosporin behandelt. Br J Dermatol 1994; 131: 713-716.
14. Kallermayer R, Szigeti R, Keeling KM, Bedekovics T, Bedwell DM. Aminoglykoside als potenzielle pharmakogenetische Wirkstoffe bei der Behandlung der Hailey-Hailey-Krankheit. J Invest Dermatol 2006; 126: 229-231.