Weltweit leiden 36 Millionen Menschen an völligem Sehverlust1. Sie können keine Formen oder sogar Lichtquellen erkennen. Bei den meisten dieser Menschen ist ihre Blindheit auf behebbare Probleme wie den Grauen Star zurückzuführen – sie haben einfach keinen Zugang zu einer angemessenen medizinischen Versorgung. Die übrigen Millionen sind jedoch aufgrund von Krankheiten erblindet, für die es derzeit keine wirksame Behandlung gibt.
„Blindheit ist eine der lebensveränderndsten Krankheiten, die ein Mensch erleben kann“, sagt William Hauswirth, Augenarzt an der Universität von Florida in Gainesville. Neben den Schwierigkeiten, die sie für die Mobilität und die Arbeitssuche mit sich bringt, werden Sehbehinderungen mit einer Reihe anderer gesundheitlicher Probleme in Verbindung gebracht, darunter Schlaflosigkeit, Angstzustände und Depressionen und sogar Selbstmordgefahr. „Die Wiederherstellung eines brauchbaren Sehvermögens würde die Lebensqualität in fast unvorstellbarer Weise verbessern“, sagt Hauswirth.
In Ländern mit hohem Einkommen, in denen vermeidbare Ursachen von Sehbehinderungen routinemäßig behandelt werden, ist die Degeneration der Netzhaut die Hauptursache für Blindheit. Dieses Gewebe, das sich im hinteren Teil des Auges befindet, enthält spezialisierte Zellen, die auf Licht reagieren und visuelle Signale verarbeiten, und ist daher entscheidend für das Sehen. Fotorezeptorzellen – Neuronen, die gemeinhin als Stäbchen und Zapfen bezeichnet werden – wandeln das auf die Netzhaut treffende Licht in elektrochemische Signale um. Diese Signale durchlaufen dann ein komplexes Netzwerk anderer Neuronen, darunter bipolare Zellen, amakrine Zellen und horizontale Zellen, bevor sie Neuronen erreichen, die als retinale Ganglienzellen bekannt sind. Die langen Fortsätze oder Axone dieser Zellen bilden den Sehnerv, über den die Signale von der Netzhaut zum visuellen Kortex des Gehirns geleitet werden, wo sie als Bilder interpretiert werden.
Netzhauterkrankungen sind häufig mit dem Verlust von Photorezeptorzellen verbunden, wodurch die Lichtempfindlichkeit des Auges abnimmt. Bei einigen Netzhauterkrankungen, darunter die altersbedingte Makuladegeneration (AMD), ist dieser Verlust auf den Ausfall der Epithelzellen zurückzuführen, die eine Schicht auf der Rückseite der Netzhaut bilden, die als retinales Pigmentepithel (RPE) bezeichnet wird. Das RPE hält die Photorezeptorzellen gesund, indem es toxische Nebenprodukte, die bei der Reaktion mit Licht entstehen, beseitigt und Nährstoffe bereitstellt. Bei Netzhauterkrankungen, bei denen die Photorezeptoren in gutem Zustand bleiben, ist die Hauptursache für Erblindung die Degeneration der retinalen Ganglienzellen.
Hören Sie sich eine Audioversion dieses Artikels an
Ihr Browser unterstützt das Audioelement nicht.
Die Vielfalt der Ursachen von Sehbehinderungen erschwert es, Lösungen zu finden. Doch Fortschritte in verschiedenen Bereichen lassen hoffen, dass fast alle Formen von Netzhauterkrankungen behandelbar werden könnten.
Ein Ansatz besteht darin, geschädigte Augen mit funktionellen Prothesen zu ergänzen oder zu umgehen. Solche bionischen Augen können derzeit nur eine begrenzte Sehkraft wiederherstellen, aber die Forscher arbeiten weiter an der Verbesserung der Fähigkeiten dieser Geräte. Eine andere Möglichkeit ist die Gentherapie. Sie steht bereits Menschen mit bestimmten genetischen Mutationen zur Verfügung, aber die Forscher versuchen, diesen Ansatz auf mehr Menschen und Krankheiten auszuweiten. Einige Wissenschaftler verfolgen auch Behandlungen auf der Grundlage einer verwandten Technik, der Optogenetik, bei der Zellen genetisch verändert werden, um die Lichtempfindlichkeit der Netzhaut wiederherzustellen. Diese Arbeiten befinden sich noch in einem frühen Stadium, aber die Forscher hoffen, dass dieser Ansatz letztendlich einem breiten Spektrum von Menschen helfen kann, da er unabhängig von den Ursachen der Netzhautdegeneration ist. Und die Bemühungen, verlorene oder geschädigte Zellen der Netzhaut entweder vor Ort oder durch Zelltransplantationen zu ersetzen, deuten darauf hin, dass selbst Netzhauterkrankungen im Spätstadium irgendwann behandelbar werden könnten.
Viele dieser Forschungen stecken noch in den Kinderschuhen. Aber Hauswirth ist zuversichtlich, was die bereits erzielten Fortschritte betrifft. Vor zehn Jahren, sagt er, musste er den Patienten oft sagen, dass er nichts für sie tun könne. „
Bionische Augen
Vor fast 30 Jahren begann Mark Humayun, ein biomedizinischer Ingenieur an der University of Southern California in Los Angeles, die Netzhaut von Blinden elektrisch zu stimulieren. In Zusammenarbeit mit Kollegen von Second Sight Medical Products, einem Medizintechnikunternehmen in Sylmar, Kalifornien, zeigten seine Experimente, dass eine solche Stimulation die visuelle Wahrnehmung von Lichtpunkten, so genannten Phosphenen, hervorrufen kann. Nachdem ein Jahrzehnt lang an Tieren gearbeitet wurde, um herauszufinden, wie viel elektrischer Strom gefahrlos an das Auge angelegt werden kann, und mit einem erheblich erweiterten Wissen über die Anzahl und die Arten von Zellen, die in degenerierenden menschlichen Netzhäuten fortbestehen, war Humayuns Team bereit, die Arbeit mit Menschen aufzunehmen. Zwischen 2002 und 2004 implantierten die Forscher sechs Menschen, die auf einem Auge vollständig oder fast vollständig erblindet waren, jeweils ein bionisches Auge – die erste Studie dieser Art. Die Empfänger des Argus I genannten Geräts berichteten, dass sie in der Lage waren, Phosphene, Richtungsbewegungen und sogar Formen wahrzunehmen2. Etwa 300 Menschen erleben nun die Welt durch das Nachfolgegerät Argus II, das 2011 von den europäischen Aufsichtsbehörden zur Verwendung bei Menschen mit Retinitis pigmentosa zugelassen wurde – einer Gruppe seltener genetischer Erkrankungen, die zur Degeneration der Sehzellen führen. Die US-amerikanische Food and Drug Administration (FDA) folgte zwei Jahre später.

Das Argus II-Implantat besteht aus einer Elektrodenanordnung, die an der Oberfläche der Netzhaut angebracht wird.Credit: Ringo Chiu/ZUMA /Alamy
Um ein Argus II-Implantat zu erhalten, müssen sich die Patienten einer Operation unterziehen, bei der ein Chip mit einem Elektroden-Array auf der Oberfläche der Netzhaut angebracht wird. Um mit dem Gerät „sehen“ zu können, überträgt eine Miniatur-Videokamera, die an einer Brille angebracht ist, Signale an eine Verarbeitungseinheit, die vom Empfänger getragen wird. Der Prozessor wandelt die Signale in Anweisungen um, die drahtlos an das implantierte Gerät übertragen werden. Die Elektroden stimulieren dann die retinalen Ganglienzellen an der Vorderseite der Netzhaut. Die Verwendung der Prothese ist ein Lernprozess. Die Empfänger müssen ihr Gehirn darauf trainieren, die neue Art der empfangenen Informationen zu interpretieren. Und da die Videokamera die Bewegung des Auges nicht verfolgt, müssen sie auch lernen, ihren Kopf zu bewegen, um ihren Blick zu lenken.
Das Gerät bietet nur eine eingeschränkte Sicht. Die Benutzer können Lichtquellen und Objekte mit kontrastreichen Kanten wie Türen oder Fenster erkennen, und einige können große Buchstaben entziffern. Diese Einschränkungen ergeben sich zum Teil daraus, dass die 60 Elektroden des Geräts im Vergleich zu den Millionen von Photorezeptorzellen in einem gesunden Auge eine sehr geringe Auflösung bieten. Aber selbst diese minimale Verbesserung kann das Leben der Menschen erheblich verbessern.
Während es sich bei Argus II um ein epiretinales Implantat handelt – das heißt, es liegt auf der Oberfläche der Netzhaut -, sind andere in der Entwicklung befindliche Geräte so konzipiert, dass sie unter der Netzhaut angebracht werden können. Diese subretinalen Implantate können Zellen stimulieren, die näher an den Zellen liegen, die normalerweise Signale an die Netzhaut weiterleiten – die Photorezeptorzellen. Durch die Stimulierung von Zellen weiter oben in der Sehbahn hoffen die Forscher, mehr von der Signalverarbeitung zu erhalten, die von einer gesunden Netzhaut durchgeführt wird.
Retina Implant, ein Biotechnologieunternehmen mit Sitz in Reutlingen, hat ein subretinales Implantat entwickelt, das Photodioden (Halbleiterbauelemente, die Licht in elektrischen Strom umwandeln) enthält, die das ins Auge einfallende Licht direkt erfassen. Dies macht eine externe Videokamera überflüssig und ermöglicht es dem Benutzer, seinen Blick ganz natürlich zu richten. Die Stromversorgung erfolgt über ein tragbares Gerät, das über eine Spule unter der Haut oberhalb des Ohrs implantiert wird. Alpha AMS, die aktuelle Version des Systems, hat in Europa die Zulassung für die Verwendung bei Menschen mit Retinitis pigmentosa erhalten.
Pixium Vision in Paris testet ein photovoltaisches subretinales Implantat namens Prima. Das System projiziert Signale von einer Videokamera, die an einer Brille angebracht ist, in das Auge. Dabei wird Nahinfrarotlicht verwendet, dessen Wellenlänge die Fotodioden im Gerät optimal ansteuert, um die Netzhautzellen zu stimulieren. Die Projektion von Bildern auf diese Weise gibt dem Benutzer eine gewisse Kontrolle über die Richtung seines Blicks, da er die Szene nur mit seinen Augen erkunden kann. Die Stromversorgung erfolgt ebenfalls durch Nahinfrarotlicht, wodurch das Implantat kabellos und die Operation zu seiner Anpassung weniger kompliziert ist. „Die Patienten lernen, wie sie schneller wieder sehen können, und die Auflösung scheint besser zu sein“, sagt José-Alain Sahel, Augenarzt an der Universität von Pittsburgh, Pennsylvania, der das Gerät bei zehn AMD-Patienten auf seine Sicherheit hin untersucht. „
Alle diese Geräte funktionieren nur, wenn noch funktionierende Zellen in der Netzhaut vorhanden sind. Bei häufigen Augenkrankheiten, die hauptsächlich Photorezeptorzellen betreffen, wie Retinitis pigmentosa und AMD, sind in der Regel noch einige Zellen vorhanden, die stimuliert werden können. Wenn jedoch zu viele retinale Ganglienzellen absterben, wie es bei fortgeschrittener diabetischer Retinopathie und Glaukom der Fall ist, können solche Implantate nicht helfen. Für Menschen ohne verbleibende Netzhautfunktion, sei es aufgrund einer Krankheit oder einer Verletzung, könnte ein alternativer bionischer Ansatz sinnvoller sein.
Humayun und seine Kollegen arbeiten an einem System, das das Auge umgeht, indem es Signale direkt an das Gehirn sendet. Die Idee ist nicht neu: In den 1970er Jahren zeigte der US-amerikanische Biomedizintechniker William Dobelle, dass die direkte Stimulation der Sehrinde die Wahrnehmung von Phosphenen3 auslöst. Aber die bionische Augentechnologie holt erst jetzt auf. Second Sight hat Orion entwickelt, ein System, das laut Humayun „im Grunde eine modifizierte Argus II“ ist. Ähnlich wie das Original verwendet es eine Videokamera und einen Signalprozessor, die drahtlos mit einem Implantat kommunizieren, wobei der Chip jedoch auf der Oberfläche des visuellen Kortex und nicht auf der Netzhaut platziert wird. Das Gerät wird derzeit an fünf Personen getestet, die aufgrund einer Augenverletzung oder einer Schädigung der Netzhaut oder des Sehnervs nur eingeschränkt oder gar nicht Licht wahrnehmen können. „Bislang sind die Ergebnisse gut“, sagt er. „
Da ein Teil der Technologie bereits an Menschen erprobt ist, ist Humayun optimistisch, dass das System in einigen Jahren die Zulassung erhalten könnte. „Natürlich ist die Hirnchirurgie mit einem anderen Risiko verbunden, aber das Verfahren ist ziemlich einfach, und Orion könnte viel mehr Patienten helfen“, sagt er. Über die Stimulierung des Gehirns zur Erzielung einer brauchbaren Sehkraft ist jedoch noch viel weniger bekannt. „Wir wissen viel über die Netzhaut, aber sehr wenig über den Kortex“, sagt Botond Roska, Neurobiologe am Institut für Molekulare und Klinische Ophthalmologie Basel in der Schweiz. „Aber wir werden nie genug wissen, wenn wir es nicht versuchen“, sagt er.
Gentherapie
Das Auge ist ein ideales Ziel für die Gentherapie. Da es relativ geschlossen ist, sollten die Viren, mit denen die Gene in die Zellen der Netzhaut eingeschleust werden, nicht in andere Teile des Körpers gelangen können. Und da das Auge ein immunprivilegierter Ort ist, ist es unwahrscheinlich, dass das Immunsystem dort eine Abwehr gegen ein solches Virus aufbaut.
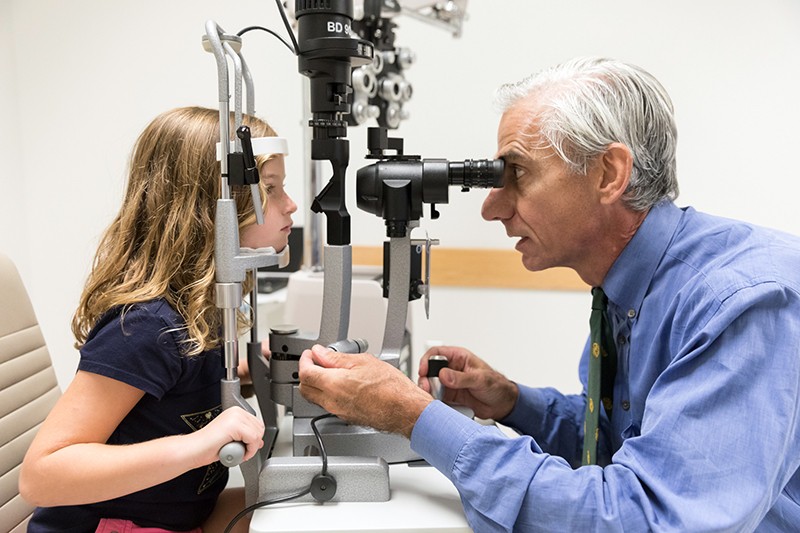
Ophthalmologe Albert Maguire untersucht die Augen eines Mädchens mit Leber kongenitaler Amaurose, dessen Sehkraft durch die Gentherapie Voretigene neparvovec (Luxturna) wiederhergestellt wurde.Credit: Children’s Hospital of Philadelphia
Erstmals haben drei Forscherteams das Potenzial der Gentherapie bei der Bekämpfung von Blindheit unter Beweis gestellt und Menschen mit Leberscher kongenitaler Amaurose (LCA) mit dieser Technik erfolgreich behandelt. Diese Erbkrankheit führt zu einer schweren Sehbehinderung, die in den ersten Lebensjahren beginnt und sich häufig als Nachtblindheit äußert, bevor sie zu einem umfassenden Sehverlust führt, der an der Peripherie des Gesichtsfelds beginnt. Etwa 1 von 40.000 Babys ist davon betroffen.
Die Forscher haben sich mit einer speziellen Form der Erkrankung befasst, die als LCA 2 bekannt ist und durch Mutationen im RPE65-Gen verursacht wird, einem Gen, das vom RPE exprimiert wird. Das mutierte Gen beeinträchtigt die Funktion des RPE, was wiederum die Photorezeptorzellen schädigt. Im Jahr 2008 zeigten die drei Teams, darunter eines unter der Leitung von Hauswirth, in frühen klinischen Versuchen, dass die Einbringung einer gesunden Kopie von RPE65 in die Netzhaut sicher war und zu einer begrenzten Verbesserung der Sehkraft führte4,5,6. Eine klinische Phase-III-Studie unter der Leitung von Albert Maguire, einem Augenarzt an der University of Pennsylvania in Philadelphia, zeigte im August 2017, dass Menschen mit LCA 2, die die Behandlung erhielten, besser in der Lage waren, bei verschiedenen Beleuchtungsstärken durch Hindernisparcours zu navigieren, als diejenigen, die nicht behandelt wurden7. Im Dezember 2017 genehmigte die FDA die Behandlung mit Voretigene neparvovec (Luxturna) und gab damit als erste Gentherapie für eine Erkrankung grünes Licht für den klinischen Einsatz.
Die Behandlung von LCA 2 ist möglich, weil die beteiligten Genmutationen rezessiv vererbt werden. Das bedeutet, dass beide Kopien von RPE65 einer Person die entsprechenden Mutationen tragen müssen, um die Erkrankung zu verursachen. Die Versorgung mit einer einzigen, nicht mutierten Version behebt daher das Problem. Bei Erkrankungen, die durch dominant vererbte Mutationen verursacht werden, ist jedoch nur eine mutierte Kopie eines Gens erforderlich, um sich zu manifestieren. In den meisten dieser Fälle hilft es nicht, einfach eine normale Kopie des Gens hinzuzufügen; stattdessen muss das mutierte Gen inaktiviert werden. Stattdessen muss das mutierte Gen inaktiviert werden. Eine Möglichkeit besteht darin, es zum Schweigen zu bringen, indem man spezifische RNA-Moleküle hinzufügt, die die Anweisungen des mutierten Gens für die Herstellung des fehlerhaften Proteins abfangen, und dann eine normale Kopie des Gens bereitstellt, um dessen Aufgaben zu übernehmen – ein Ansatz, der als Unterdrückung und Ersatz bezeichnet wird. Eine andere Möglichkeit besteht darin, die Mutation mit Hilfe der Gen-Editing-Technik CRISPR-Cas9 zu korrigieren. Forscher der Universität Modena und Reggio Emilia in Modena, Italien, demonstrierten diesen Ansatz 2016 an einem Mausmodell für Retinitis pigmentosa8. Im darauffolgenden Jahr korrigierte ein Team in den Vereinigten Staaten damit die Mutation, die eine Art von Glaukom verursacht, sowohl bei Mäusen als auch in kultivierten menschlichen Zellen9.
Ein wichtiger Motor für den Fortschritt in der Gentherapie war die Verwendung von Adeno-assoziierten Viren (AAV), um Ersatzgene in Zellen einzubringen. AAVs haben sich als sicher erwiesen, unter anderem, weil sie nicht in das Genom ihrer Wirtszelle integriert werden, was das Risiko einer Krebsentstehung minimiert. Außerdem können sie sich aufgrund ihrer geringen Größe weit im Auge ausbreiten und so eine große Anzahl von Zellen infizieren. Die Fähigkeit von AAVs, Gene zu übertragen, hat jedoch ihre Grenzen: Einige Gene sind einfach zu groß für AAVs, darunter ABCA4, dessen Mutationen zur Stargardt-Krankheit, einer vererbbaren Form der Makuladegeneration, führen können. Zwei Umgehungsmöglichkeiten werden derzeit verfolgt. Bei der ersten wird ein Virus mit einer größeren Übertragungskapazität, wie z. B. ein Lentivirus, verwendet, um Ersatzgene zu übertragen. Die Sicherheit und Wirksamkeit dieses Ansatzes ist nicht bekannt, aber klinische Versuche sind im Gange. Eine zweite Strategie besteht darin, das Ersatzgen in zwei Hälften zu zerlegen und jede Hälfte separat in die Zelle zu transportieren, zusammen mit einer Möglichkeit, sie neu zu kombinieren. „Das funktioniert im Moment zumindest in einem Tiermodell“, sagt Hauswirth.
Ungeachtet des Ansatzes hat die Gentherapie eine erhebliche Einschränkung. Mehr als 250 Gene sind an der Erblindung beteiligt, und da jedes von ihnen von zahlreichen Arten von Mutationen betroffen sein kann, ist die Zahl der potenziellen therapeutischen Ziele enorm. So führen beispielsweise mehr als 100 Mutationen im Gen RHO zu Retinitis pigmentosa, der häufigsten dominant vererbten Netzhauterkrankung. Die Entwicklung einer Gentherapie für jede einzelne Mutation ist nicht praktikabel, sagt Hauswirth.
Forscher arbeiten an einer möglichen Lösung, die den Ansatz des Unterdrückens und Ersetzens abwandelt. Anstatt auf RHO-Kopien mit einer bestimmten Mutation abzuzielen, verwenden sie eine Silencing-RNA, um die gesamte Expression des Gens zu unterdrücken, unabhängig davon, ob RHO mutiert ist oder nicht, während sie eine Ersatzkopie liefern, die immun gegen die Silencing-RNA ist. Ein Team unter der Leitung von Jane Farrar, einer Genetikerin am Trinity College in Dublin, zeigte 2011, wie vielversprechend diese Strategie in einem Mausmodell für dominante Retinitis pigmentosa10 ist. Im Jahr 2018 testeten Hauswirth und Kollegen den Ansatz an Hunden mit Retinitis pigmentosa11. Sie zeigten, dass die Degeneration von Photorezeptorzellen in behandelten Bereichen der Netzhaut aufgehalten werden konnte – eine Verbesserung, die mindestens acht Monate lang anhielt. Mit dieser Strategie werden alle Mutationen, die eine dominant vererbte Retinitis pigmentosa verursachen können, mit einer einzigen Behandlung angegangen, so dass die Gentherapie von rezessiven auf dominant vererbte Erkrankungen „auf recht einfache Weise“ ausgedehnt werden kann, sagt Hauswirth. Er plant, zu untersuchen, wie gut Hunde, die die Behandlung erhalten haben, in einem Labyrinth navigieren können, und sammelt die Sicherheitsdaten, die für den Beginn einer klinischen Studie erforderlich sind.
Optogenetik
Die Gentherapie funktioniert nur bei Menschen, deren Blindheit durch eine genetische Mutation verursacht wird. Sie eignet sich auch nicht für die Behandlung von Netzhauterkrankungen im Endstadium, bei denen nicht mehr genügend Zellen für eine Reparatur vorhanden sind. Ein ähnlicher Ansatz, der auf einer Technik namens Optogenetik beruht, ist jedoch nicht krankheitsanfällig und könnte zu Behandlungen für verschiedene Stadien der Degeneration führen. Bei der Optogenetik werden Gene, die Zellen in die Lage versetzen, lichtempfindliche Proteine, so genannte Opsine, zu produzieren, durch einen Virus übertragen. Die Einführung von Opsinen kann die Lichtempfindlichkeit geschädigter Photorezeptoren bis zu einem gewissen Grad wiederherstellen oder sogar andere Zellen der Netzhaut, einschließlich bipolarer Zellen oder retinaler Ganglienzellen, lichtempfindlich machen.

Mit Hilfe der Optogenetik konnte die Lichtempfindlichkeit der Zapfenzellen (grün) in einem Mausmodell der Retinitis pigmentosa wiederhergestellt werden; der Erfolg der Technik wurde durch die Messung der Aktivität einer retinalen Ganglienzelle (magenta) beurteilt, die von den Zapfen als Reaktion auf Licht stimuliert wird.Credit: IOB.ch
Problematisch ist jedoch, dass die Photorezeptorzellen des Auges mit einem breiten Spektrum von Lichtintensitäten zurechtkommen – sie funktionieren sowohl bei hellem Sonnenlicht als auch in der Dämmerung gut -, während die Opsine einen begrenzten Bereich haben und oft bei hohen Lichtintensitäten besser funktionieren. Eine mögliche Lösung ist ein System, das ähnlich wie das bionische Augensystem Prima von Pixium Vision funktioniert, bei dem die Empfänger mit einer Brille ausgestattet sind, die eine Videokamera enthält, die die Sicht des Benutzers aufnimmt, und einen Projektor, der auf das Auge gerichtet ist. Wie bei Prima besteht der Vorteil darin, dass die Art des Lichts, das in das Auge gelangt, auf die Modifikation der Netzhaut zugeschnitten werden kann. In diesem Fall werden jedoch die Intensität und die Wellenlänge gewählt, die die neu eingeführten Opsine am besten ansteuern, und nicht die implantierten Photodioden.
GenSight Biologics, ein Biotechnologieunternehmen in Paris, zu dessen Gründern Sahel und Roska gehören, testet bereits ein solches System. Es will den Ganglienzellen der Netzhaut ein Opsin zuführen, aber es gibt einen möglichen Haken: Ganglienzellen der Netzhaut sind von Natur aus lichtempfindlich. Sie exprimieren Melanopsin, ein Protein, das am Pupillen-Lichtreflex beteiligt ist, bei dem sich die Pupille des Auges als Reaktion auf helles Licht verengt. Um dies zu vermeiden, verwenden die Forscher von GenSight ein Opsin, das auf rote Lichtwellenlängen anspricht, da Melanopsin bevorzugt auf Licht am blauen Ende des Spektrums reagiert. Das Unternehmen hat im Oktober 2018 eine klinische Studie im Frühstadium bei Menschen mit fortgeschrittener Retinitis pigmentosa begonnen, die nur noch über ein minimales Sehvermögen verfügen. An der Studie nehmen Kohorten aus dem Vereinigten Königreich, Frankreich und den Vereinigten Staaten teil, und die ersten Ergebnisse werden bis Ende 2020 erwartet.
„Dies ist ein einfacher Ansatz, und wir müssen sehen, was er bringt“, sagt Roska. „Dann können wir zu immer ausgefeilteren Ansätzen übergehen.“ Ein Problem besteht darin, dass viele der Erkrankungen, die mit optogenetischen Techniken behandelt werden könnten, mit der Degeneration bestimmter Teile der Netzhaut einhergehen, während in anderen Bereichen das Sehvermögen erhalten bleibt. Das Licht, das die Opsine antreibt, ist sichtbar und könnte das verbleibende natürliche Sehvermögen beeinträchtigen. In Zukunft könnten Opsine, die auf nahes Infrarotlicht reagieren, es ermöglichen, dass optogenetische Behandlungen mit dem verbleibenden natürlichen Sehvermögen einhergehen.
Zellregeneration
Die Stammzelltherapie könnte Blindheit sogar im Spätstadium der Krankheit heilen. Da Stammzellen dazu gebracht werden können, sich in jede Art von Zelle zu verwandeln, könnten sie verwendet werden, um neue Netzhautzellen zu züchten, die in das Auge transplantiert werden, um die verlorenen Zellen zu ersetzen. Studien an Tieren haben jedoch gezeigt, dass nur ein kleiner Teil der transplantierten Neuronen in der Lage ist, sich korrekt in die komplexen neuronalen Schaltkreise der Netzhaut zu integrieren. Dies stellt ein erhebliches Hindernis für Stammzellbehandlungen dar, die darauf abzielen, Neuronen in der Netzhaut zu ersetzen.

Die komplexe Zellstruktur der Netzhaut umfasst Schichten von Photorezeptoren (grün) sowie Blutgefäße und Nerven (magenta).Credit: Louise Hughes/SPL
Die Zellen, aus denen das Pigmentepithel der Netzhaut besteht, liegen dagegen außerhalb des Schaltkreises der Netzhaut. Stammzellbasierte Therapien sind daher besonders vielversprechend bei Erkrankungen wie AMD und Retinitis pigmentosa, bei denen die RPE-Zellen degenerieren. „Die Photorezeptoren müssen mit dem Schaltkreis verbunden sein, das retinale Pigmentepithel jedoch nicht“, sagt Roska. „Hier ist man am nächsten dran, Fortschritte zu machen.“ Zunächst versuchten die Forscher, die Netzhaut mit aus Stammzellen gewonnenen RPE-Zellen in Suspension zu injizieren, aber es blieben zu wenige dort, wo sie gebraucht wurden. Mehrere Teams sind nun der Ansicht, dass ein besserer Ansatz darin besteht, RPE-Zellen in Form einer vorgeformten Schicht ins Auge zu transplantieren, die dann von einem biokompatiblen Gerüst in Position gehalten wird. „
Im März 2018 gab das London Project to Cure Blindness – eine Zusammenarbeit zwischen dem University College London und dem Moorfields Eye Hospital in London – die Ergebnisse einer Phase-I-Studie bekannt, bei der zwei Menschen mit feuchter AMD (einer seltenen, schweren Form der AMD, bei der es zu abnormem Wachstum und Undichtigkeiten der Blutgefäße kommt) eine Platte aus RPE-Zellen in die Netzhaut implantiert wurde. Beide Patienten haben den Eingriff gut vertragen und konnten 21-29 Buchstaben mehr auf einer Lesetafel lesen als vor der Behandlung12. Im darauf folgenden Monat berichtete ein Team unter der Leitung von Humayun über ähnliche Phase-I-Ergebnisse bei fünf Personen mit trockener AMD, der häufigeren Form der Erkrankung13. Diese ersten Ergebnisse sind sehr vielversprechend. „Das hat zu großer Aufregung geführt“, sagt Humayun. Die Ergebnisse müssen jedoch durch Phase-III-Studien mit einer größeren Anzahl von Teilnehmern bestätigt werden, und Humayun warnt, dass die Behandlung noch viele Jahre von einer klinischen Anwendung entfernt sein könnte, da bisher noch keine Stammzelltherapie für eine Netzhauterkrankung den Zulassungsprozess durchlaufen hat.
Ein verwandter Ansatz, der sich noch im Anfangsstadium der Grundlagenforschung befindet, könnte die Hoffnung erfüllen, verloren gegangene Neuronen zu ersetzen und damit die Tür zu Behandlungen für eine Vielzahl von Augenkrankheiten zu öffnen. Beim Menschen teilen sich reife Neuronen nicht und können sich daher nicht regenerieren, was die Wiederherstellung des Sehvermögens besonders schwierig macht. Dies gilt jedoch nicht für alle Tiere. Reptilien und bestimmte Fische können Netzhautneuronen regenerieren, und auch Vögel verfügen über eine gewisse Regenerationsfähigkeit. Thomas Reh, ein Neurowissenschaftler an der University of Washington in Seattle, versucht, diese Fähigkeit beim Menschen zu nutzen. Anstatt jedoch im Labor gezüchtete Zellen zu transplantieren, will Reh Zellen, die sich bereits in der Netzhaut befinden, dazu bringen, sich in neue Neuronen zu differenzieren.
Im Jahr 2001 schlug Reh vor, dass Müller-Glia – Zellen, die die Netzhaut strukturieren und ihre Funktion unterstützen – die Quelle neuer Neuronen sind, die bei Fischen und Vögeln beobachtet worden waren14. Er und sein Team machten sich daraufhin daran herauszufinden, ob Müller-Glia zur Erzeugung neuer Neuronen in Mäusen genutzt werden können. Im Jahr 2015 veränderten sie Mäuse so, dass sie Ascl1 produzierten, ein Protein, das für die Bildung von Neuronen in Fischen wichtig ist, und schädigten dann die Netzhaut der Tiere15. Sie hofften, dass Ascl1 die Müller-Glia dazu veranlassen würde, sich in Neuronen zu verwandeln.
Bei erwachsenen Mäusen gelang es nicht, neue Neuronen zu produzieren, bei jungen Mäusen hingegen schon. Nikolas Jorstad, Biochemiker und Doktorand in Rehs Team, schlug vor, dass chemische Veränderungen, die während der Entwicklung am Chromatin (einem Komplex aus DNA, RNA und Proteinen) im Zellkern vorgenommen werden, in reifen Zellen den Zugang zu Genen blockieren könnten, die es Müller-Glia ermöglichen, sich in Neuronen zu verwandeln. Im August 2017 zeigte das Team um Reh, dass es durch die Einführung eines Enzyms, das solche Veränderungen rückgängig macht, Müller-Glia zur Differenzierung bewegen kann16. „Zum ersten Mal konnten wir Neuronen in der erwachsenen Maus regenerieren“, sagt Reh. „Nach all den Jahren war ich ziemlich aufgeregt.“ Obwohl es sich nicht um echte Photorezeptorzellen handelte und sie eher wie bipolare Zellen aussahen, schlossen sich die Neuronen an die bestehenden Schaltkreise an und waren lichtempfindlich. „Ich war überrascht, dass sie sich so gut verbinden“, sagt Reh.
Obwohl die Arbeit noch lange nicht bereit ist, Netzhauterkrankungen bei Menschen zu behandeln, hat sie ein großes Potenzial. Der nächste Schritt wird darin bestehen, die Studien an Tieren mit Augen zu wiederholen, die denen des Menschen ähnlicher sind. Das Team von Reh arbeitet bereits mit Netzhautzellkulturen von nicht-menschlichen Primaten. Die Forscher müssen auch herausfinden, wie sie den Differenzierungsprozess steuern können, um bestimmte Zelltypen wie Stäbchen und Zapfen zu produzieren. „Jetzt, wo wir einen Fuß im Geschäft mit der Herstellung von Neuronen haben, wären Zapfen großartig“, sagt Reh.
Wenn der Ansatz erfolgreich ist, könnte er allgemein anwendbar sein. „Letztendlich wird dies die Art und Weise sein, wie all diese Augenkrankheiten behandelt werden“, prophezeit Reh. „Es macht einfach Sinn. Man muss sich nicht darum kümmern, dass die Transplantation richtig erfolgt. Die Zellen sind genau da, wo man sie braucht.“
Humayun ist von der Arbeit ebenfalls ermutigt. „Ich freue mich über jeden, der eine neue gute Idee hat“, sagt er. „Es ist noch sehr früh, es ist ein hohes Risiko, aber sag niemals nie. Das ist es, was ich gelernt habe.“