- Introducción
- Materiales y Métodos
- Preparación de las muestras
- Preparación de bibliotecas MinION y secuenciación
- Análisis genómico
- Resultados
- Datos de secuenciación MinION y ensamblaje de genomas virales
- Extensión y origen de la contaminación cruzada de las muestras
- Discusión
- Contribuciones de los autores
- Financiación
- Declaración de conflicto de intereses
- Agradecimientos
- Material complementario
- Notas al pie
Introducción
La secuenciación metagenómica tiene el potencial de permitir la identificación imparcial de patógenos a partir de una muestra clínica. Es prometedor que sirva como ensayo único y universal para el diagnóstico de enfermedades infecciosas directamente a partir de muestras sin necesidad de conocimientos a priori (Bibby, 2013; Miller et al., 2013; Schlaberg et al., 2017). Además de la identificación de especies patógenas, los datos de secuencias metagenómicas amplias y profundas podrían proporcionar información relevante para determinar el tratamiento y el pronóstico, detectar brotes y hacer un seguimiento de la epidemiología de la infección (Greninger et al., 2010; Yang et al., 2011; Qin et al., 2012; Loman et al., 2013). Las plataformas de secuenciación de próxima generación (NGS) pueden producir un rendimiento masivo de datos a un coste modesto, sin embargo, su aplicación en el diagnóstico clínico y la salud pública se ha visto limitada por la complejidad, la lentitud y la inversión de capital.
El MinION es un secuenciador genómico de una sola molécula en tiempo real del tamaño de la palma de la mano desarrollado por Oxford Nanopore Technologies (ONT). El tamaño compacto y la naturaleza en tiempo real del MinION podrían facilitar la aplicación de la secuenciación metagenómica en las pruebas en el punto de atención para las enfermedades infecciosas, como lo demuestran varios estudios de prueba de concepto, incluyendo la identificación de Chikungunya (CHIKV), Ébola (EBOV), y el virus de la hepatitis C (HCV) a partir de muestras de sangre clínica humana sin enriquecimiento de objetivos (Greninger et al, 2015), y la detección de patógenos bacterianos a partir de muestras de orina (Schmidt et al., 2016) y muestras respiratorias, sin necesidad de un cultivo previo (Pendleton et al., 2017).
El rendimiento de datos de MinION ha aumentado enormemente desde su lanzamiento en 2015, y cada celda de flujo consumible genera ahora hasta 10-20 Gb de datos de secuencias de ADN. Esto permite a los usuarios hacer un uso más eficiente de la celda de flujo (y reducir el coste) mediante la multiplexación de varias muestras en una sola ejecución de secuenciación. ONT ha desarrollado conjuntos de códigos de barras sin PCR que permiten el multiplexado de hasta 12 muestras.
La detección del virus de la gripe A en múltiples muestras respiratorias podría ser un uso diagnóstico de un ensayo de secuenciación MinION multiplexado. Sin embargo, al secuenciar directamente a partir de muestras con una potencial amplia gama de títulos virales, es importante ser consciente de la posibilidad de contaminación cruzada de la muestra, tanto durante la preparación de la biblioteca como en la etapa de demultiplexación del código de barras bioinformático después de la secuenciación. Aquí presentamos un conjunto de datos de secuenciación MinION único y los resultados de la investigación sobre el alcance y el origen de la contaminación por códigos de barras cruzados en la secuenciación multiplex.
Materiales y Métodos
Utilizamos una muestra de lavado nasal de hurón infectada con el virus de la gripe A como ejemplo y también se añadieron dos alícuotas de muestras de lavado nasal negativas de hurones no infectados (existencias preexistentes no utilizadas de un estudio no relacionado) con los virus del dengue y chikungunya por separado. Ninguno de estos virus es relevante para el diagnóstico clínico en muestras respiratorias, pero actúan aquí como marcadores claros y distintos para la evaluación de la contaminación cruzada de las muestras. Las bibliotecas de secuenciación de cada muestra se prepararon en paralelo, junto con un control de lavado nasal negativo, con código de barras, y se secuenciaron individualmente. A continuación, se agrupó una alícuota de las bibliotecas de secuenciación y se realizó la secuenciación MinION multiplex. A continuación, se analizaron las lecturas de las cuatro ejecuciones individuales (denominadas «CHIKV», «DENV», «FLU-A» y «Negative») y la ejecución multiplex (denominada «Multiplexed») para investigar el alcance y la fuente de la contaminación cruzada de las muestras.
Preparación de las muestras
La licencia del proyecto fue revisada por el AWERB local (Animal Welfare and Ethics Review Board) y posteriormente fue concedida por el Ministerio del Interior. Se extrajo el ARN, utilizando el kit de ARN viral QIAamp (Qiagen) de acuerdo con las instrucciones del fabricante, de lavados nasales de hurones que contenían el virus de la gripe A (H1N1) (A/California/04/2009) y de un conjunto de muestras de lavados nasales negativos. A las alícuotas del extracto de la muestra negativa se les añadió el ARN viral del dengue (DENV) (cepa TC861HA, GenBank: MF576311) o del CHIKV (cepa S27, GenBank: MF580946.1) de la colección nacional de virus patógenos1. Las muestras se trataron con DNasa utilizando TURBO DNase (Thermo Fisher Scientific, Waltham, MA, Estados Unidos) y se purificaron utilizando el kit RNA Clean & ConcentratorTM-5 (Zymo Research). Se preparó el ADNc y se amplificó utilizando un método de amplificación de un solo rimero independiente de la secuencia (Greninger et al., 2015) modificado como se describió anteriormente (Atkinson et al., 2016). El ADNc amplificado se cuantificó utilizando el kit de ensayo Qubit dsDNA HS (Thermo Fisher Scientific, Waltham, MA, Estados Unidos), y se utilizó 1 μg como entrada para cada preparación de la biblioteca MinION, con la excepción del control negativo en el que se utilizó toda la muestra (32 ng).
Preparación de bibliotecas MinION y secuenciación
Se utilizó el kit de secuenciación de ligación 1D (SQK-LSK108) y el kit de código de barras nativo 1D (EXP-NBD103) de acuerdo con los protocolos estándar de la ONT, con la excepción de que solo se incluyó un código de barras en cada una de las cuatro preparaciones de bibliotecas. Cada biblioteca se ejecutó en una celda de flujo individual y se hizo una quinta biblioteca agrupada combinando las cuatro bibliotecas con códigos de barras individuales. Las bibliotecas se secuenciaron en celdas de flujo R9.4. El diseño del estudio se muestra en la Figura 1.
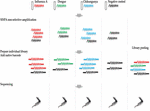
Figura 1. Resumen del diseño del estudio. Se extrajo el ARN de cuatro muestras, incluyendo una muestra de lavado nasal de hurón infectado con el virus de la gripe A, dos muestras de lavado nasal de hurón negativas con virus del dengue y chikungunya, y un control de lavado nasal de hurón negativo. Se preparó el ADNc y se amplificó utilizando un método de amplificación de un solo rimero independiente de la secuencia. Las bibliotecas de secuenciación de cada muestra se prepararon en paralelo, con código de barras, y se secuenciaron en celdas de flujo individuales. La secuenciación multiplex también se realizó agrupando las cuatro bibliotecas individuales. Se analizaron las lecturas de las cuatro ejecuciones individuales y de la ejecución multiplex para evaluar el alcance y el origen de la contaminación por códigos de barras cruzados en la secuenciación multiplex.
Análisis genómico
Las lecturas se marcaron de forma básica utilizando Albacore v2.1.7 (ONT) con demultiplexación de códigos de barras. Las lecturas de cada corrida de secuenciación se mapearon a las secuencias genómicas de cada virus usando Minimap2 (Li, 2018). El número de lecturas mapeadas a la referencia se contó usando Pysam2. El ensamblaje de novo se realizó utilizando Canu v1.7 (Koren et al., 2017), y el borrador del genoma resultante se pulió utilizando Nanopolish (Mongan et al., 2015) con los datos de nivel de señal.
Para permitir el demultiplexado estricto del código de barras de los datos de secuenciación MinION multiplex, realizamos dos rondas de análisis utilizando Porechop (v0.2.23). La presencia de una secuencia adaptadora en medio de una lectura es una firma de quimera. Utilizamos Porechop para examinar cada lectura y las que tenían una región central que compartía >75% de identidad con la secuencia del adaptador fueron identificadas como lecturas quiméricas. En Porechop, establecimos la opción «-middle_threshold» y elegimos un umbral de 75. En la segunda ronda, utilizamos Porechop para buscar la secuencia del código de barras tanto al principio como al final de una lectura; las lecturas se asignaron sólo si se encontraba el mismo código de barras en los dos extremos. Establecimos la opción «-require_two_barcodes» en Porechop y fijamos el umbral para la puntuación del código de barras en 70. Para encontrar la firma potencial de las lecturas quiméricas, examinamos las señales de corriente de las lecturas almacenadas en el archivo FAST5 por el secuenciador MinION. Las señales de corriente se extrajeron utilizando ONT fast5 API4 y se trazaron utilizando ggplot2 implementado en R5 para una comparación de lecturas quiméricas y no quiméricas.
Resultados
El rendimiento de cada ejecución de secuenciación MinION varió debido a las diferencias en el tiempo de ejecución. Se alcanzó un número máximo de ∼2,4 M de lecturas en la ejecución de secuenciación multiplexada y en la ejecución individual de CHIKV, debido a los mayores tiempos de ejecución (Tabla suplementaria S1). Las lecturas del virus pinchado representaron el 96% de los datos en las series individuales de secuenciación del CHIKV y el DENV, y el 78% para la muestra FLU-A (Tabla 1). El porcentaje de lecturas virales dentro de cada muestra con código de barras en los datos de la secuenciación multiplexada se aproxima al de los datos de las muestras ejecutadas individualmente (Tabla 2). Cada genoma viral tenía una profundidad media de cobertura ultra alta (>8.000) en los datos de secuenciación individual y multiplexada, y el ensamblaje de novo fue capaz de recuperar genomas casi completos para los tres virus con un 99.9% de identidades en comparación con la referencia de GenBank.

Tabla 1. Resumen de los resultados de mapeo y ensamblaje de novo para los datos de la secuenciación MinION de bibliotecas individuales.

Tabla 2. Resumen de los resultados del mapeo y del ensamblaje de novo para los datos de la secuenciación MinION multiplex.
Extensión y origen de la contaminación cruzada de las muestras
Cada muestra fue codificada por barras, y secuenciada tanto individualmente como multiplexada, lo que nos permitió examinar el rendimiento de la demultiplexación del código de barras de Albacore. En los datos de las muestras secuenciadas individualmente, cabría esperar la presencia de un único código de barras nativo. En las series de secuenciación individual de CHIKV (código de barras NB01), DENV (NB09) y FLU-A (NB10), descubrimos que 86, 109 y 17 lecturas, respectivamente, se asignaron a intervalos de códigos de barras que no se esperaba que estuvieran presentes en la biblioteca (lo que representa el 0,0036, el 0,0129 y el 0,001% de las lecturas totales). En los datos de secuenciación múltiple, se asignaron 41 lecturas (0,0016%) a códigos de barras no incluidos en los experimentos (es decir, un código de barras distinto de NB01, NB05, NB09 o NB10). Las definimos como lecturas mal asignadas (Figura 2A).
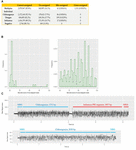
Figura 2. (A) resumen del número y porcentaje de lecturas correctamente asignadas, no asignadas, mal asignadas y cruzadas en cada ejecución de secuenciación. Las no asignadas se refieren a las lecturas que Albacore no puede asignar a ningún intervalo debido a una puntuación de código de barras inferior a 60, las mal asignadas se refieren a las lecturas que se asignaron a intervalos de código de barras no incluidos en este experimento y las asignadas de forma cruzada se refieren a las lecturas que se asignaron a intervalos de código de barras incorrectos; (B) distribución de las puntuaciones de código de barras notificadas por Albacore para las lecturas mal asignadas y las asignadas de forma cruzada en los datos de secuenciación múltiple; (C) comparación de la señal bruta de una lectura quimérica y una lectura correctamente asignada. La señal de la lectura quimérica posee una señal de estancamiento y una enorme señal de pico en el centro de la lectura.
Para examinar la posible contaminación de laboratorio en la preparación de la biblioteca de secuenciación, se mapearon todas las lecturas de cada ejecución individual contra las secuencias genómicas de los tres virus. No se encontró ninguna lectura que se originara en un genoma preparado en una biblioteca diferente, lo que sugiere que no hubo contaminación in vitro. La biblioteca de secuenciación múltiple se preparó agrupando las bibliotecas individuales no contaminadas tras la ligadura del código de barras y el adaptador. Sin embargo, los resultados del mapeo muestran que 1.311 (0,0543%) lecturas se asignaron al genoma diana incorrecto, lo que implica que se asignaron de forma cruzada a los bines de código de barras incorrectos (posteriormente denominados «lecturas de asignación cruzada»), a pesar de que la biblioteca de secuenciación multiplexada se agrupó con las bibliotecas individuales no mostraron ninguna lectura de asignación cruzada. Se planteó la hipótesis de que las lecturas mal asignadas y cruzadas se debían a una puntuación baja del código de barras, y se investigaron las puntuaciones del código de barras de estas lecturas. La mayoría de las lecturas mal asignadas tenían una puntuación de código de barras <70, sin embargo, las lecturas con asignación cruzada tenían puntuaciones más diversas que iban desde 60 hasta casi 100 (Figura 2B). Este resultado sugiere que las lecturas mal asignadas y las cruzadas proceden de fuentes diferentes. Comparamos las lecturas cruzadas con una pequeña base de datos que incluía las secuencias genómicas de los tres virus incluidos en este estudio, y demostramos que 1074/1311 (82%) de estas lecturas podían alinearse de forma cruzada con más de un genoma viral (1.047 lecturas) o alinearse de forma cruzada con regiones distintas dentro del mismo genoma (27 lecturas), lo que sugiere que son quimeras. Para confirmar esta observación, investigamos las señales de corriente brutas de algunas lecturas de alineación cruzada en comparación con las de las lecturas correctamente asignadas (Figura 2C). Las señales de corriente de una lectura correctamente asignada suelen incluir (i) una señal de poro abierto de alta corriente que representa el momento en que el poro de secuenciación cambia de un adaptador a otro, (ii) una señal de estancamiento, que se refiere al periodo de tiempo en que una secuencia de ADN está en el poro pero aún no se mueve, y (iii) la traza de la señal de secuenciación del ADN. Por el contrario, una lectura quimérica posee una señal de estancamiento y una enorme señal de pico en el centro de la lectura. Las lecturas quiméricas pueden poseer dos secuencias de código de barras diferentes al principio y al final, lo que confunde la asignación de una casilla de código de barras. En conjunto, estos datos demuestran dos categorías de error que contribuyen a la contaminación cruzada de muestras en nuestro conjunto de datos: (i) lecturas quiméricas (representan ∼80% de todas las lecturas de asignación cruzada); (ii) lecturas con baja puntuación de código de barras. Para mejorar la calidad de nuestro conjunto de datos final, exploramos el impacto de diferentes enfoques de demultiplexación de códigos de barras para eliminar las lecturas con asignación cruzada (Tabla 3). El filtrado de las lecturas que poseen un adaptador interno puede eliminar el 90% de las lecturas con asignación cruzada y perder el 24% de las lecturas totales. También probamos un esquema de filtrado más estricto que requería dos códigos de barras (uno al principio y otro al final de la lectura) para hacer una asignación. Este enfoque eliminó todas las lecturas con asignación cruzada excepto dos, pero perdió el 56% del total de lecturas.

Tabla 3. Eliminación de lecturas cruzadas y pérdida de datos de secuenciación totales mediante dos enfoques de filtrado utilizando Porechop.
También investigamos el alcance de las posibles lecturas quiméricas en los datos de secuenciación. Para las secuencias individuales de CHIKV, DENV y FLU-A, los resultados del mapeo muestran que el 2,3, el 3,0 y el 2,7% de las lecturas mapeadas, respectivamente, poseen una alineación suplementaria y se alinean al menos dos veces con el mismo genoma (Tabla 4). Consideramos tanto las lecturas clasificadas por el código de barras como las no clasificadas en los datos de secuenciación múltiple. Los resultados muestran que el 2,0% de las lecturas mapeadas poseen alineación suplementaria y se alinearon al menos dos veces con el mismo genoma, mientras que el 0,052% de las lecturas totales se alinearon con al menos dos genomas distintos.

TABLE 4. Resumen del número y porcentaje de lecturas no quiméricas, autoquiméricas y cruzadas en cada corrida de secuenciación.
Discusión
El objetivo final de nuestra investigación es desarrollar un ensayo de diagnóstico basado en la secuenciación metagenómica de nanoporos que permita realizar pruebas en el punto de atención para enfermedades infecciosas. La secuenciación multiplex ofrece la oportunidad de mejorar la escalabilidad y reducir el coste, sin embargo, la contaminación cruzada de las muestras puede dar lugar a errores en los datos y a una falsa interpretación de los resultados.
En este experimento, agrupamos bibliotecas limpias y realizamos la secuenciación MinION multiplex con el fin de investigar el alcance y el origen de la contaminación cruzada. Identificamos que el 0,056% de las lecturas totales se asignaron de forma cruzada a los bines de códigos de barras incorrectos, lo que es comparable a los reportados para las plataformas de secuenciación Illumina de diferentes estudios (entre el 0,06 y el 0,25%) (Nelson et al., 2014; D’Amore et al., 2016; Wright y Vetsigian, 2016). Nuestros resultados mostraron que las lecturas quiméricas son la fuente predominante de errores de asignación de códigos de barras cruzados. Las lecturas quiméricas asignadas de forma cruzada en este conjunto de datos solo podrían haberse formado durante la secuenciación y no durante la preparación de las bibliotecas, ya que estaban completamente ausentes en los datos de secuenciación de las bibliotecas individuales, y el único paso de procesamiento posterior fue mezclar las bibliotecas de secuenciación finales antes de cargarlas. Tenemos la hipótesis de que el algoritmo actual implementado en Albacore no puede reconocer la corta disociación entre las secuencias de ADN que corren simultáneamente a través del nanoporo, concatenando así más de una secuencia en el mismo archivo Fast5.
Las lecturas quiméricas se observaron en los datos de secuenciación MinION antes en White et al. (2017). A través de los análisis de los datos de secuenciación MinION de tres amplicones de interferón diferentes, los autores encontraron que el 1,7% de las lecturas mapeadas eran quimeras. Nuestros hallazgos se suman al conocimiento que apoya que las quimeras son comunes en los datos de secuenciación MinION. Identificamos que entre el 2 y el 3% del total de lecturas en tres datos de secuenciación individual y uno multiplex son quimeras. Nuestro estudio difiere de los trabajos anteriores en los dos aspectos siguientes. En primer lugar, aportamos pruebas directas de que pueden formarse lecturas quiméricas después de la preparación de la biblioteca y durante la secuenciación; además, relacionamos estas quimeras con la contaminación de muestras cruzadas en la secuenciación MinION múltiplex, como se ha comentado anteriormente. Por otro lado, la configuración de nuestro experimento tiene limitaciones para identificar posibles quimeras formadas en la preparación de la biblioteca, en particular durante el paso de ligación del adaptador en el protocolo estándar de secuenciación multiplex. En segundo lugar, nuestros resultados reflejan el estado actual de la secuenciación MinION porque utilizamos el kit de secuenciación ONT más nuevo y representativo, incluyendo el kit de secuenciación de ligadura 1D (SQK-LSK108) y el kit de código de barras nativo 1D (EXP-93 NBD103). La tecnología de secuenciación por nanoporos se está desarrollando rápidamente y se está mejorando en todos los aspectos. Por ejemplo, se ha lanzado un nuevo kit de secuenciación por ligadura de ADN (SQK-LSK109) y un kit de secuenciación directa de ARN (SQK-RNA001); se ha mejorado el algoritmo de llamada base implementado en Albacore y Guppy. Todos estos cambios tienen un efecto sobre la extensión de la quimera en los datos de secuenciación Nanopore y la contaminación por códigos cruzados durante la secuenciación multiplex. La limitación de este estudio fue el pequeño número de experimentos, el trabajo adicional utilizando diferentes configuraciones de experimentos añadiría a nuestra comprensión de los datos de secuenciación múltiple de Nanopore. Además, es importante investigar las contribuciones de los factores potenciales a la contaminación de códigos de barras cruzados, lo que arrojaría luz sobre las mejores prácticas para analizar los datos de secuenciación múltiplex.
En resumen, nuestro estudio demostró que las lecturas quiméricas son la fuente predominante de errores de asignación de códigos de barras cruzados en la secuenciación MinION múltiplex. Destaca la necesidad de un filtrado cuidadoso de los datos de secuenciación MinION multiplex antes del análisis posterior, y el compromiso entre la sensibilidad y la especificidad que se aplica a los métodos de demultiplexación de códigos de barras.
Contribuciones de los autores
SP, KL, SL y YX realizaron la secuenciación MinION. YX analizó los datos. Todos los autores diseñaron el estudio, participaron en la interpretación de los resultados y en la redacción del manuscrito, y leyeron y aprobaron la versión final de este manuscrito.
Financiación
Este trabajo fue apoyado por el NIHR Oxford Biomedical Research Centre.
Declaración de conflicto de intereses
Los autores declaran que la investigación se llevó a cabo en ausencia de cualquier relación comercial o financiera que pudiera interpretarse como un potencial conflicto de intereses.
Agradecimientos
Nos gustaría dar las gracias al Dr. Anthony Marriott (Public Health England) por proporcionar aspirados nasales de hurón.
Material complementario
Notas al pie
.