Muchos de los métodos de visualización e interpretación de los datos de expresión génica pueden utilizarse tanto para los experimentos de microarrays como para los de RNA-seq. Algunos de los métodos más comunes se discuten a continuación.
Mapas de calor y clustering
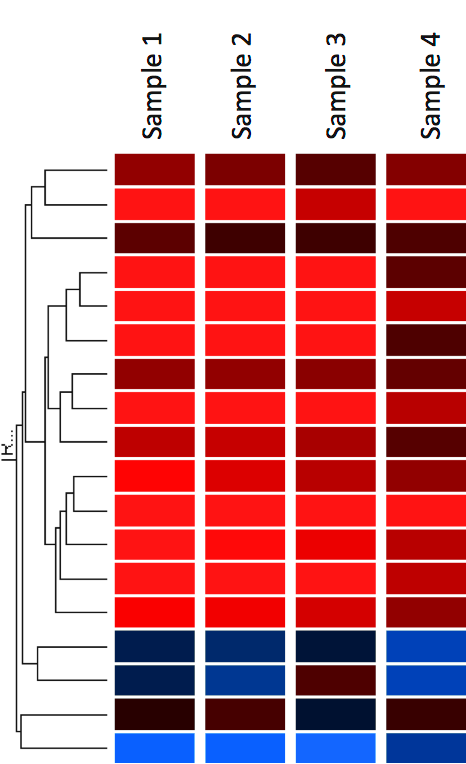
Un método común para visualizar los datos de expresión génica es mostrarlos como un mapa de calor (Figura 12). El mapa de calor también puede combinarse con métodos de clustering que agrupan genes y/o muestras en función de la similitud de su patrón de expresión génica. Esto puede ser útil para identificar genes que están comúnmente regulados, o firmas biológicas asociadas con una condición particular (por ejemplo, una enfermedad o una condición ambiental) (4).
En los mapas de calor los datos se muestran en una cuadrícula donde cada fila representa un gen y cada columna representa una muestra. El color y la intensidad de los cuadros se utilizan para representar los cambios (no los valores absolutos) de la expresión génica. En el ejemplo siguiente, el rojo representa los genes regulados al alza y el azul representa los genes regulados a la baja. El negro representa la expresión sin cambios.
Análisis de enriquecimiento de conjuntos de genes y análisis de vías
Un enfoque común para interpretar los datos de expresión génica es el análisis de enriquecimiento de conjuntos de genes basado en la anotación funcional de los genes expresados diferencialmente (Figura 13). Esto es útil para averiguar si los genes expresados diferencialmente están asociados a un determinado proceso biológico o función molecular.
La Ontología de Genes, que contiene una anotación estandarizada de los productos de los genes, se utiliza comúnmente para este propósito. Funciona comparando la frecuencia de las anotaciones individuales en la lista de genes (por ejemplo, los genes expresados diferencialmente) con una lista de referencia (normalmente todos los genes del microarray o del genoma). El enriquecimiento de las vías biológicas suministradas por KEGG, Ingenuity, Reactome o WikiPathways puede realizarse de forma similar (12,13).
Las herramientas populares para el enriquecimiento de conjuntos de genes y el análisis de vías biológicas incluyen:
- DAVID (herramienta online gratuita)
- GSEA (gratuito)
- Ingenuity (requiere licencia)
- Reactome (gratuito)

Análisis de redes
El análisis de redes es complementario al análisis de vías y puede utilizarse para mostrar cómo interactúan los componentes clave de diferentes vías. Esto puede ser útil para identificar eventos reguladores que influyen en múltiples procesos y vías biológicas (12,13).