INTRODUCCIÓN
Las enfermedades pulmonares intersticiales forman un grupo sumamente heterogéneo de trastornos que tienen en común la existencia de un componente inflamatorio del parénquima pulmonar que, eventualmente, puede evolucionar hacia la fibrosis y que provocan, desde el punto de vista funcional, una alteración restrictiva que puede acabar conduciendo a la insuficiencia respiratoria.
Aunque son enfermedades cuyo diagnóstico y manejo se lleva a cabo en el ámbito especializado de la neumología, el tratamiento se sigue, fundamentalmente, de modo ambulatorio, y dado que éste puede y suele verse salpicado por diversas complicaciones, es importante que el médico de atención primaria conozca las pautas actuales con que se enfoca el tratamiento de estas enfermedades, ya que en no pocas ocasiones se verá consultado por pacientes con este tipo de problemas, ya sea por complicaciones respiratorias o de otro tipo.
La finalidad de esta revisión es hacer una actualización de las modalidades terapéuticas que hoy en día se pueden ofrecer a estos pacientes. Creemos que es de la mayor importancia no sólo el conocimiento actualizado de este tema por el médico de atención primaria, sino también la colaboración con el neumólogo de cara a conseguir un buen cumplimiento terapéutico del paciente y una detección y tratamiento precoz de las complicaciones que puedan presentarse.
Dentro de las enfermedades intersticiales, la más relevante por su frecuencia y por su gravedad es la fibrosis pulmonar idiopática (FPI). Por otro lado, su tratamiento es el modelo en el que se basa el de la práctica totalidad de estas enfermedades, a excepción hecha de aquellas en las que puede encontrarse un agente etiológico conocido, ya que en éstas la eliminación de dicho agente será la base fundamental de la terapéutica. Por ello, nos vamos a centrar en la revisión de las pautas actuales de manejo de la FPI. Haremos primero un breve recuerdo de sus características clínicas para luego desarrollar pormenorizadamente el manejo actual de la misma.
DESCRIPCIÓN CLÍNICA
La FPI representa probablemente el prototipo más agresivo de las neumopatías intersticiales difusas. Su prevalencia se ha cifrado por algunos autores en 5 casos por 100.000 habitantes1, aunque en realidad es desconocida. Su patogenia se relaciona con alteraciones en las complejas vías del sistema inmunológico2,3. Por este motivo, la mayoría de los fármacos utilizados son agentes inmunosupresores.
El modo de presentación clínico más habitual es la disnea de inicio insidioso acompañada de tos seca, que puede llegar a ser el síntoma que más molesto resulte al paciente por su carácter paroxístico y disneizante. Por lo general, la disnea es al inicio poco llamativa y es necesario un elevado índice de sospecha para diagnosticarla de forma precoz. Ocasionalmente, la evolución es rápida desde el inicio y conduce en pocos meses a la insuficiencia respiratoria irreversible, tal y como fue característico en las descripciones iniciales de Hamman y Rich. Sin embargo, lo más habitual es que el cuadro haya cursado meses de evolución para cuando el paciente hace su primera consulta.
A la exploración física son muy característicos los crepitantes finos y secos (velcro rales) que pueden auscultarse en más de un 80% de los casos. En cuadros avanzados puede apreciarse disnea de pequeños esfuerzos o incluso cianosis labial. En aproximadamente un 30-50% de los casos pueden observarse acropaquias. La sospecha de FPI se ve reforzada cuando en la radiografía de tórax y en la TC aparecen signos como patrón reticular, áreas de infiltración en vidrio deslustrado y disminución de volumen pulmonar. Por último, la exploración funcional confirmará la existencia de un patrón restrictivo y la gasometría arterial la existencia de hipoxemia. Es de gran valor en casos dudosos la demostración de hipoxemia al esfuerzo.
La confirmación definitiva del diagnóstico sólo la ofrece la biopsia pulmonar. Sin embargo, la realidad es que incluso en muchas de las series publicadas predominan los casos sin diagnóstico histológico. Recientemente, la Sociedad Americana del Tórax (ATS) ha propuesto que la existencia de todos estos criterios permiten, con bastante probabilidad, y en ausencia de exposición a fármacos, elementos ambientales o enfermedades del tejido conectivo, poder llegar al diagnóstico de FPI en ausencia de biopsia pulmonar4 en casos en los que la realización de ésta suponga un riesgo inaceptable.
La neumonía intersticial usual (NIU) es el patrón histopatológico que identifica a estos pacientes4. Aquellos con patrones de neumonía intersticial descamativa, enfermedad intersticial asociada a bronquiolitis respiratoria, neumonía intersticial inespecífica, neumonía intersticial linfoide, neumonía intersticial aguda, y bronquiolitis obliterante con neumonía organizada se consideran entidades diferentes.
Una vez establecido el diagnóstico y la gravedad clínica, la mayoría de pacientes requieren tratamiento porque es una enfermedad fatal y sin remisión espontánea. La respuesta al tratamiento es escasa. La supervivencia a los 5 años es del 50%5,2,6-8. El tratamiento requiere unos 3-6 meses antes de que se establezca su efectividad. Los informes sobre el tratamiento de estas enfermedades son escasos: ensayos terapéuticos no controlados que en su mayoría incluyen un pequeño número de pacientes9.
A pesar de la inevitable progresión insidiosa e irreversible de la enfermedad, no se justifica el nihilismo terapéutico. Existen ejemplos de fibrosis no tratada con lenta progresión que desemboca en una fibrosis irreversible no respondedora a tratamiento o que desarrolla una fase rápidamente progresiva. Probablemente, todos requieren tratamiento en el momento de su presentación. No obstante, aquellos pacientes con contraindicaciones para el tratamiento o los que no tienen evidencia de alteración funcional pueden seguir un programa de seguimiento con intervalos de 3-6 meses. También es difícil el manejo de pacientes ancianos con histología de fibrosis predominante y con alteraciones graves de la función pulmonar. Estos pacientes reciben un tratamiento breve de 3-6 meses para determinar la reversibilidad. Pocos de ellos van a tener una respuesta positiva.
A menudo es difícil determinar si el tratamiento produce el efecto deseado. La mejoría subjetiva sucede a menudo (hasta en el 70% de los pacientes). La mejoría objetiva se da en un 20-30%. Los parámetros más utilizados para definir una mejoría funcional (objetiva) son el aumento de la capacidad pulmonar total (TLC) o de la capacidad vital (CV), un aumento de la capacidad de difusión y una reducción o normalización del descenso de la SaO2 durante el ejercicio4,10-12.
TRATAMIENTO
Corticoides
Los corticoides son la base fundamental del tratamiento de la FPI, si bien el éxito que con ellos se obtiene es más bien escaso: una respuesta favorable ocurre en una tercera parte de los pacientes. El fundamento de su uso es suprimir la alveolitis crónica. Se cree que actúan suprimiendo la migración de neutrófilos y linfocitos en el pulmón, al disminuir la concentración de inmunocomplejos, alterando la función de los macrófagos alveolares y la adhesión de neutrófilos a las superficies endoteliales.
No está claro por qué algunos pacientes responden y otros no. Puede estar relacionado con la presencia de receptores de superficie en las células del parénquima pulmonar. Un tiempo corto de evolución de los síntomas, la alteración funcional y radiológica leve, el sexo femenino y la juventud de los pacientes parecen asociarse a una mejor expectativa.
Sin embargo, y pese a ser el tratamiento más utilizado, su rendimiento a largo plazo no ha sido confirmado por ningún estudio prospectivo, realizado a ciegas y con un grupo control que utilice placebo.
Izumi et al analizaron la supervivencia de 222 pacientes y constataron que a los 10 años no había diferencia en la supervivencia entre los que habían recibido esteroides en comparación con los no tratados13. Pese a ello, ante un paciente con FPI que presente un trastorno de su función pulmonar o sintomatología que interfiera con el desarrollo de su vida habitual, es razonable iniciar un intento de tratamiento con corticoides. De hecho, la mejoría espontánea de la enfermedad es muy rara, y si se decide retrasar el inicio del tratamiento debe ser a expensas de mantener un estrecho control del paciente que permita iniciarlo en cuanto se evidencie un deterioro del proceso.
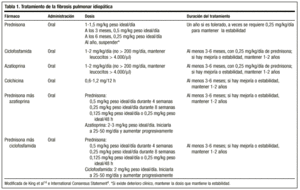
El tratamiento clásico comienza con prednisona 1,5-2 mg/kg peso ideal/día en una dosis única por la mañana, sin exceder la dosis de 100 mg. Tras 2-3 meses se revalúa al paciente con radiografía y estudio funcional. Si es respondedor se desciende la prednisona (1-2 mg/semana) hasta una dosis de mantenimiento de 0,5 mg/kg peso ideal/día, que podrá reducirse posteriormente. Si no existe respuesta se añade un fármaco inmunosupresor. En general, es poco probable que se pueda suspender el tratamiento, y se suelen mantener con dosis de 0,25 mg/kg peso ideal/día.
Recientemente, las Sociedades Americana y Europea del Tórax, con el objeto de evitar los efectos secundarios de este fármaco y obtener mejores tasas de supervivencia, han alcanzado un consenso para iniciar el tratamiento con una combinación de prednisona u otro corticoide a dosis equivalentes y un inmunosupresor (azatioprina o ciclofosfamida)4: prednisona 0,5 mg/kg peso ideal/día durante 4 semanas; 0,25 mg/kg peso ideal/día durante 8 semanas, y descender entonces a 0,125 mg/kg peso ideal/día o 0,25 mg/kg peso ideal/48 h.
No existen guías sobre la duración del tratamiento; el tiempo mínimo es de un año. Es posible que tras meses o años la enfermedad se reactive, y en esa circunstancia hay que iniciar el tratamiento con los mismos agentes. En todo caso, la respuesta al tratamiento debe ser valorada siempre en términos de mejoría objetiva, y la posibilidad de mantener al paciente crónicamente con una dosis que garantice su estabilidad se reservará para aquellos casos en los que haya mejoría con corticoides, deterioro al disminuirlos y nueva mejoría al volver a aumentar la dosis. La dosis que necesitará cada paciente sólo puede determinarse individualmente en función de la respuesta a los cambios de la misma. Muchos autores consideran que el tratamiento necesario es de por vida14.
Cuando la enfermedad es inicialmente grave se ha propuesto utilizar dosis altas de corticoides. Keogh et al han utilizado altas dosis intermitentes de corticoides parenterales en pacientes con afección grave: metilprednisolona intravenosa, 2 g una vez a la semana, más prednisona oral, 0,25 mg/kg/día. El fundamento es que tienen un efecto más pronunciado sobre la alveolitis neutrofílica (asociada a mal pronóstico) que los esteroides a bajas dosis15. No obstante, este trabajo adolece de algunos defectos, como su seguimiento corto de 6 meses, el poco número de pacientes y las dosis de prednisona menores que las estándares9. Un tratamiento inicial con altas dosis de corticoides intravenosos (metilprednisolona 250 mg/6 h) se ha utilizado en un esfuerzo para controlar la enfermedad lo más precozmente posible cuando ésta es rápidamente progresiva. No existen estudios controlados que determinen si estos métodos son efectivos; sin embargo, las dosis más altas parecen tener mayor eficacia, aunque los efectos secundarios son también mayores14.
AGENTES CITOTOXICOS
Se han utilizado clásicamente como alternativa a los corticoides cuando éstos no eran eficaces, y también cuando sus efectos secundarios impiden que se puedan seguir administrando. Siguiendo el espíritu eminentemente práctico de este artículo, vamos a revisar aquellos agentes que han demostrado su utilidad en la práctica clínica, aunque, como en el caso de los corticoides, carecemos de series largas, prospectivas y aleatorias que permitan conocer con exactitud cuál es su papel exacto en el tratamiento de la FPI.
Azatioprina
Su mecanismo de acción se basa en la sustitución de las purinas en la síntesis del ácido desoxirribonucleico, con lo que inhibe la adenindeaminasa e inactiva los linfocitos. Otros efectos son la supresión de la actividad de las células natural killer, producción de anticuerpos y la citotoxicidad celular dependiente de anticuerpos.
La dosis recomendada es de 2 mg/kg/día, sin exceder 200 mg/día, durante un período de 3-6 meses. Su administración se inicia a dosis de 50 mg/día y, si es bien tolerada, se va incrementando la dosis a intervalos de 1 o 2 semanas hasta alcanzar la dosis planeada.
Sus efectos secundarios fundamentales son de orden hematológico: leucopenia, anemia, trombocitopenia, aplasia de células rojas y pancitopenia. Es necesario monitorizar el recuento de leucocitos, y si éste baja de 4.000/μl hay que suspender la azatioprina hasta que se remonte dicha cifra. Además, pueden aparecer molestias gastrointestinales, como náuseas y vómitos, úlcera péptica, diarrea y también una elevación leve de las enzimas hepáticas. En total, hasta en un 20-30% de los casos, los efectos secundarios obligan a retirar la azatioprina.
Raghu et al compararon la eficacia de los glucocorticoides a altas dosis frente a azatioprina asociada a glucocorticoides a dosis bajas. La utilización de este fármaco se acompañó de una moderada mejoría de los parámetros funcionales respiratorios y de un aumento en la supervivencia17.
Ciclofosfamida
Es un agente alquilante. Su modo de acción es la depleción de linfocitos y, por tanto, suprime su función. La dosis recomendada es de 2 mg/kg/día única, oral, generalmente considerada con 0,25 mg/kg/día de prednisona oral, sin exceder los 200 mg/día, como mínimo, durante 3 meses y hasta 9-12 meses.
El efecto secundario más importante es la leucopenia. Se debe mantener un recuento total de leucocitos mayor de 3.000/μl o de neutrófilos mayor de 1.500/μl. Otras complicaciones incluyen trombocitopenia, hematuria secundaria a cistitis hemorrágica, anorexia, náuseas y vómitos, supresión de la médula ósea, azoospermia y amenorrea, infección y enfermedad maligna hematológica.
Kolb et al estudiaron 18 pacientes con FPI con el objeto de determinar la eficacia y seguridad del tratamiento con pulsos de ciclofosfamida. Ésta se administró de forma intermitente (1-1,3 g/mes) junto a prednisolona oral durante un año. Tras este período se suspendió la ciclofosfamida y se continuó con las dosis alcanzadas de corticoides8. Se les siguió un mínimo de 14 meses, controlando la CV, la TLC, el FEV1, la capacidad de transferencia de CO (DLCO), la SaO2, así como los síntomas. En un paciente tuvo que suspenderse el tratamiento por neumonías de repetición; 11 pacientes se consideraron respondedores (5 mejoraron y 6 se estabilizaron) y 6 se deterioraron. La toxicidad a largo plazo de la ciclofosfamida depende de las dosis acumuladas. Las dosis intermitentes son igual de efectivas y mejor toleradas que las diarias. Los respondedores presentaban un mayor porcentaje de linfocitos en el BAL. Otro dato a tener en cuenta es que la CV inicial de los respondedores fue significativamente mejor debido a un menor tiempo de evolución, lo que parece indicar que este tratamiento se debería comenzar precozmente.
Baughman y Lower trataron a 33 pacientes con FPI con 1.000-1.800 mg de ciclofosfamida/2 semanas durante 18 meses. Aquellos que sobrevivieron > 18 meses presentaron una mejoría significativa en la FVC que se mantuvo durante el año siguiente. La reducción de la prednisolona fue posible de 32 a 4 mg/día16.
Penicilamina
Agente antifibrosante. Se ha sugerido que al interferir con la formación de las uniones covalentes intra e intermoleculares de las fibras de colágena inhibe el desarrollo de la fibrosis pulmonar. Se han descrito algunos casos de enfermedades de tejido conectivo con fibrosis pulmonar asociada en los que ha tenido cierta eficacia, pero no hay estudios controlados en series grandes de pacientes, por lo que no puede considerarse actualmente como una alternativa válida de tratamiento. Se requieren más estudios, pues, para poder hacer alguna recomendación a este respecto.
Colchicina
Disminuye la secreción de colágeno y aumenta la actividad colagenolítica. También tiene un efecto antiinflamatorio. Se requieren estudios para determinar de forma más precisa su papel en el tratamiento de esta enfermedad pero existen trabajos en los que se le atribuye una eficacia similar a la de los corticoides y, además, sus efectos secundarios no son graves. Se recomienda a dosis de 0,6 mg, una o dos veces al día, en pacientes refractarios a corticoides, sola o en combinación con agentes inmunosupresores. Aunque hay autores que la sitúan en primera línea de tratamiento, actualmente carecemos de datos para establecerla como tal (tabla 1).
TRASPLANTE PULMONAR
Los casos en los que la enfermedad progresa sin respuesta al tratamiento y acaba provocando insuficiencia respiratoria son, desgraciadamente, los más numerosos en esta enfermedad. No hay en la actualidad ninguna perspectiva de tratamiento médico para estos pacientes excepto el paliativo. En este panorama desolador, surgió en la década de los ochenta la posibilidad de trasplante pulmonar, que se ha desarrollado hasta constituir en la actualidad una opción terapéutica que debe considerarse como una herramienta más de uso clínico y que ya no se limita al campo de lo experimental. Su indicación más idónea se realiza en personas menores de 65 años, con ausencia de enfermedad progresiva e irreversible en otros órganos, así como de infección activa no resuelta o infección por gérmenes multirresistentes. Asimismo, es necesaria una predisposición personal y un estado nutricional adecuados. Los pacientes con una CPT menor del 60% del esperado o una capacidad de difusión pulmonar menor del 40% tienen una supervivencia menor de 2 años. En este grado de disfunción precisan oxigenoterapia y desaturan fácilmente con el ejercicio mínimo. En estos casos debería ser valorado el trasplante pulmonar, en cuanto se hace necesaria la oxigenoterapia y se verifica el progreso imparable de la enfermedad. El tipo de trasplante de elección es el unipulmonar, reservando el bipulmonar a los casos en que existan dudas sobre el comportamiento del pulmón residual. Cuando el paciente tiene menos de 50 años y padece una insuficiencia cardíaca derecha, se puede plantear el trasplante cardiopulmonar. Sin embargo, en esta situación la enfermedad está muy avanzada y es probable que no soporte la espera18,19. Las expectativas de supervivencia postrasplante se cifran en un 80% al primer año y en un 50-60% a los 5 años, lo que mejora ostensiblemente el pronóstico que tiene un paciente con FPI que ha llegado a la insuficiencia respiratoria.
EVALUACION DE LA RESPUESTA AL TRATAMIENTO
En ausencia de complicaciones, pueden transcurrir hasta 6 meses antes de obtener algún tipo de respuesta al tratamiento. Pasado este tiempo, si el paciente ha mejorado o está estable, se deben mantener las mismas dosis. Si ha empeorado, hay que cambiar a otra terapia alternativa (otro agente inmunosupresor o incluso el trasplante). A los 12 meses se debe revaluar de la misma forma y a partir de los 18 meses individualizar según la respuesta. Se recomienda continuar indefinidamente sólo en aquellos pacientes con mejoría continua o estabilización (tabla 2).

Una respuesta favorable se define por dos o más de los siguientes criterios en dos visitas consecutivas:
1. Mejoría de los síntomas, sobre todo en el grado de esfuerzo requerido para que el paciente deba parar cuando hace un esfuerzo o un descenso en la frecuencia o gravedad de la tos.
2. Reducción de las alteraciones radiológicas.
3. Mejoría funcional definida por dos o más de los siguientes parámetros:
Aumento >= 10% en la TLC o CV (o >= 200 ml).
Aumento >= 15% en DLCO o >= 3 ml/min/mmHg.
Mejoría o normalización de la SaO2 o pO2 (aumento >= 4% o >= 4 mmHg) durante un prueba de esfuerzo cardiorrespiratoria.
Una respuesta estable (también considerada favorable) se define por dos o más de los siguientes criterios en dos visitas consecutivas:
1. Cambio de
2. Cambio de
3. Sin cambios en la SaO2 o pO2 (aumento
Un fracaso del tratamiento se define por dos o más de los siguientes criterios en dos visitas consecutivas:
1. Aumento de los síntomas, sobre todo disnea o tos.
2. Aumento de las alteraciones radiológicas, especialmente imágenes en panal o signos de hipertensión pulmonar.
3. Deterioro funcional definido por dos o más de los siguientes parámetros:
Descenso >= 10% en la TLC o CV (o >= 200 ml).
Descenso >= 15% en DLCO o >= 3 ml/min/mmHg.
Empeoramiento de la SaO2 (descenso >= 4%) o elevación del gradiente alvéolo-arterial (AapO2) (>= 4 mmHg) en reposo o durante una prueba de esfuerzo cardiorrespiratoria.
CONCLUSIONES
Podemos afirmar que la fibrosis pulmonar idiopática es una enfermedad fatal, sin remisión espontánea y con escasa respuesta al tratamiento. Los estudios sobre este último aspecto son escasos y cuentan con un pequeño número de pacientes. Los corticoides mantenidos un mínimo de un año a diferentes dosis son la pauta más aceptada, solos o en combinación con otros agentes inmunosupresores. Recientemente, se ha preconizado por parte de las sociedades Americana del Tórax y Respiratoria Europea una terapia combinada4. Agentes antifibrosantes están siendo objeto de investigación con la intención de evitar la instauración del proceso fibrótico y, por tanto, su irreversibilidad, puesto que muchos pacientes son diagnosticados en etapas tardías. Dadas las tasas de supervivencia y la evolución se debería considerar el trasplante pulmonar tan pronto como fracasa el tratamiento de los pacientes y precisan oxigenoterapia. De todo lo comentado se desprende la necesidad de establecer estudios multicéntricos con la idea de reclutar un número suficiente de pacientes con los que poder valorar la eficacia de los tratamientos administrados, así como identificar la historia natural de la enfermedad y la incidencia y la prevalencia reales4,20. Por otro lado, se recomienda identificar e iniciar el tratamiento en las primeras fases de la enfermedad con el objeto de obtener mayores tasas de supervivencia.