Dans le monde, 36 millions de personnes ont une perte totale de la vision1. Elles ne peuvent pas voir les formes ni même les sources de lumière. Pour la plupart de ces personnes, leur cécité découle de problèmes rectifiables tels que la cataracte – elles n’ont tout simplement pas accès à des soins de santé appropriés. Les millions de personnes restantes, cependant, sont aveugles en raison d’affections pour lesquelles il n’existe actuellement aucun traitement efficace.
« La cécité est l’une des affections qui bouleversent le plus la vie d’une personne », explique William Hauswirth, ophtalmologue à l’université de Floride à Gainesville. Outre les difficultés qu’elle entraîne pour la mobilité et la recherche d’un emploi, la déficience visuelle est associée à une foule d’autres problèmes de santé, notamment l’insomnie, l’anxiété et la dépression, et même le risque de suicide. « Restaurer une vision utile apporterait une amélioration presque inimaginable de la qualité de vie », affirme Hauswirth.
Dans les pays à revenu élevé où les causes évitables de déficience visuelle sont systématiquement traitées, la principale cause de cécité est la dégénérescence de la rétine. Situé à l’arrière de l’œil, ce tissu contient des cellules spécialisées qui réagissent à la lumière et traitent les signaux visuels, et est donc crucial pour la vision. Les cellules photoréceptrices – des neurones communément appelés bâtonnets et cônes – convertissent la lumière qui frappe la rétine en signaux électrochimiques. Ces signaux passent ensuite par un réseau complexe d’autres neurones, dont les cellules bipolaires, les cellules amacrines et les cellules horizontales, avant d’atteindre des neurones appelés cellules ganglionnaires de la rétine. Les longues projections, ou axones, de ces cellules forment le nerf optique, le long duquel les signaux de la rétine sont transportés vers le cortex visuel du cerveau, où ils sont interprétés sous forme d’images.
Les troubles rétiniens impliquent généralement la perte de cellules photoréceptrices, ce qui appauvrit la sensibilité de l’œil à la lumière. Dans certains troubles rétiniens, notamment la dégénérescence maculaire liée à l’âge (DMLA), cette perte résulte d’une défaillance des cellules épithéliales qui forment une couche à l’arrière de la rétine, appelée épithélium pigmentaire rétinien (EPR). L’EPR maintient les cellules photoréceptrices en bonne santé en nettoyant les sous-produits toxiques produits lors de la réaction avec la lumière, ainsi qu’en fournissant des nutriments. Dans les troubles rétiniens où les photorécepteurs restent en bon état, la principale cause de cécité est la dégénérescence des cellules ganglionnaires de la rétine.
Écouter une version audio de cet article
Votre navigateur ne prend pas en charge l’élément audio.
La variété des causes de la déficience visuelle rend plus difficile la recherche de solutions. Mais les avancées dans plusieurs domaines font naître l’espoir que presque toutes les formes de troubles rétiniens pourraient devenir traitables.
Une approche consiste à augmenter ou à contourner les yeux endommagés avec des prothèses fonctionnelles. De tels yeux bioniques ne peuvent restaurer qu’une vision limitée pour le moment, mais les chercheurs continuent de pousser les capacités de ces dispositifs. Une autre option est la thérapie génique. Déjà accessible aux personnes présentant des mutations génétiques spécifiques, les chercheurs cherchent à étendre cette approche à un plus grand nombre de personnes et de pathologies. Certains scientifiques s’intéressent également aux traitements basés sur une technique connexe, l’optogénétique, qui consiste à modifier génétiquement des cellules pour rétablir la sensibilité de la rétine à la lumière. Ces travaux n’en sont qu’à leurs débuts, mais les chercheurs espèrent que cette approche pourra un jour aider un large éventail de personnes, car elle est indépendante des causes de la dégénérescence rétinienne. Et les efforts visant à remplacer les cellules perdues ou endommagées de la rétine, que ce soit in situ ou par des greffes de cellules, laissent entrevoir que même les troubles rétiniens à un stade avancé pourraient éventuellement devenir traitables.
Une grande partie de cette recherche n’en est qu’à ses débuts. Mais Hauswirth est optimiste quant aux progrès qui ont déjà été réalisés. Il y a dix ans, dit-il, il devait souvent dire aux patients qu’il ne pouvait rien faire pour eux. « Pour beaucoup de ces maladies, cela a totalement changé. »
Des yeux bioniques
Il y a près de 30 ans, Mark Humayun, ingénieur biomédical à l’Université de Californie du Sud à Los Angeles, a commencé à stimuler électriquement les rétines des personnes atteintes de cécité. En collaboration avec des collègues de Second Sight Medical Products, une entreprise de technologie médicale située à Sylmar, en Californie, ses expériences ont montré que cette stimulation pouvait induire la perception visuelle de taches lumineuses appelées phosphènes. Après une décennie de travaux sur des animaux pour déterminer la quantité de courant électrique pouvant être appliquée sans danger à l’œil, et forte d’une connaissance beaucoup plus approfondie du nombre et des types de cellules qui persistent dans les rétines humaines en dégénérescence, l’équipe de Humayun était prête à commencer à travailler avec des personnes. Entre 2002 et 2004, les chercheurs ont implanté un œil bionique dans chacune des six personnes atteintes de cécité totale ou presque totale d’un œil – le premier essai de ce type. Les bénéficiaires de l’appareil, connu sous le nom d’Argus I, ont déclaré pouvoir percevoir des phosphènes, des mouvements directionnels et même des formes2. Environ 300 personnes découvrent aujourd’hui le monde grâce au successeur de ce dispositif, l’Argus II, qui a été approuvé par les autorités réglementaires européennes en 2011 pour les personnes atteintes de rétinite pigmentaire – un groupe de maladies génétiques rares qui entraînent la dégénérescence des cellules photoréceptrices. La Food and Drug Administration (FDA) américaine a fait de même deux ans plus tard.

L’implant Argus II comprend un réseau d’électrodes qui est fixé à la surface de la rétine.Crédit : Ringo Chiu/ZUMA /Alamy
Pour être équipés d’un Argus II, les patients subissent une intervention chirurgicale visant à fixer une puce contenant un réseau d’électrodes à la surface de la rétine. Pour « voir » avec le dispositif, une caméra vidéo miniature montée sur une paire de lunettes relaie des signaux à une unité de traitement qui est portée par le destinataire. Le processeur convertit les signaux en instructions qui sont transmises sans fil au dispositif implanté. Les électrodes stimulent alors les cellules ganglionnaires situées à l’avant de la rétine. L’utilisation de la prothèse est un processus d’apprentissage. Les receveurs doivent entraîner leur cerveau à interpréter le nouveau type d’informations reçues. Et comme la caméra vidéo ne suit pas le mouvement de l’œil, ils doivent également apprendre à bouger leur tête pour diriger leur regard.
Le dispositif ne fournit qu’une vision limitée. Les utilisateurs peuvent détecter les sources de lumière et les objets aux bords très contrastés, comme les portes ou les fenêtres, et certains peuvent déchiffrer de grandes lettres. Ces limites résultent en partie du fait que les 60 électrodes du dispositif fournissent une résolution très faible par rapport aux millions de cellules photoréceptrices d’un œil sain. Mais même cette amélioration minime peut améliorer considérablement la vie des gens.
Alors que l’Argus II est un implant épirétinien – ce qui signifie qu’il repose à la surface de la rétine – d’autres dispositifs en cours de développement sont conçus pour être placés sous la rétine. Ces implants sous-rétiniens peuvent stimuler des cellules qui sont plus proches de celles qui introduisent normalement les signaux dans la rétine – les cellules photoréceptrices. En stimulant les cellules plus haut dans la voie visuelle, les chercheurs espèrent préserver une plus grande partie du traitement des signaux effectué par une rétine saine.
Retina Implant, une société de biotechnologie basée à Reutlingen, en Allemagne, a construit un implant sous-rétinien comprenant des photodiodes (dispositifs semi-conducteurs qui convertissent la lumière en courant électrique) qui détectent directement la lumière entrant dans l’œil. Cela élimine le besoin d’une caméra vidéo externe, permettant aux utilisateurs de diriger leur regard naturellement. L’alimentation est fournie par une unité portative, par le biais d’une bobine implantée sous la peau, au-dessus de l’oreille. Alpha AMS, la version actuelle du système, a reçu l’approbation réglementaire en Europe pour être utilisé chez les personnes atteintes de rétinite pigmentaire.
Pixium Vision à Paris teste un implant sous-rétinien photovoltaïque appelé Prima. Le système projette dans l’œil les signaux d’une caméra vidéo montée sur des lunettes en utilisant une lumière proche de l’infrarouge, dont la longueur d’onde pilote de manière optimale les photodiodes du dispositif pour stimuler les cellules rétiniennes. La projection d’images de cette manière permet à l’utilisateur de contrôler la direction de son regard, puisqu’il peut explorer la scène en ne bougeant que ses yeux. L’alimentation est également assurée par la lumière infrarouge proche, ce qui rend l’implant sans fil et l’opération chirurgicale pour le poser moins compliquée. « Les patients apprennent à retrouver la vision plus rapidement, et la résolution semble meilleure », explique José-Alain Sahel, ophtalmologue à l’université de Pittsburgh, en Pennsylvanie, qui mène des essais de sécurité du dispositif chez dix personnes atteintes de DMLA. « Il est encore tôt, mais c’est très prometteur. »
Tous ces dispositifs ne fonctionnent que lorsque des cellules fonctionnelles restent dans la rétine. Dans les pathologies oculaires courantes qui affectent principalement les cellules photoréceptrices, notamment la rétinite pigmentaire et la DMLA, il reste généralement quelques cellules à stimuler. Mais lorsque trop de cellules ganglionnaires de la rétine meurent, comme c’est le cas dans la rétinopathie diabétique avancée et le glaucome, ces implants ne peuvent rien faire. Pour les personnes sans aucune fonction rétinienne restante, que ce soit en raison d’une maladie ou d’une blessure, une autre approche bionique pourrait être plus pertinente.
Humayun et ses collègues travaillent sur un système qui contourne l’œil en envoyant des signaux directement au cerveau. L’idée n’est pas nouvelle : dans les années 1970, l’ingénieur biomédical américain William Dobelle a montré que la stimulation directe du cortex visuel déclenchait la perception de phosphènes3. Mais la technologie de l’œil bionique commence seulement à rattraper son retard. Second Sight a mis au point Orion, un système qui, selon Humayun, est « essentiellement un Argus II modifié ». Comme l’original, il utilise une caméra vidéo et un processeur de signaux qui communiquent sans fil avec un implant, mais la puce est placée à la surface du cortex visuel plutôt que sur la rétine. Le dispositif est actuellement testé sur cinq personnes dont la perception de la lumière est limitée ou nulle en raison d’une blessure à l’œil ou d’une lésion de la rétine ou du nerf optique. « Jusqu’à présent, les résultats sont bons », dit-il. « Nous ne sommes pas encore surpris par quoi que ce soit. »
Compte tenu du fait qu’une partie de la technologie est déjà éprouvée chez l’homme, Humayun est optimiste et pense que le système pourrait recevoir une autorisation réglementaire d’ici quelques années. « Évidemment, la chirurgie du cerveau présente un niveau de risque différent, mais la procédure est assez simple, et l’Orion pourrait aider beaucoup plus de patients », dit-il. Cependant, on en sait beaucoup moins sur la stimulation du cerveau pour obtenir une vision utile. « Nous en savons beaucoup sur la rétine mais très peu sur le cortex », déclare Botond Roska, neurobiologiste à l’Institut d’ophtalmologie moléculaire et clinique de Bâle, en Suisse. « Mais nous n’en saurons jamais assez si nous n’essayons pas », ajoute-t-il.
Thérapie génique
L’œil est une cible idéale pour la thérapie génique. Parce qu’il est relativement autonome, les virus utilisés pour transporter les gènes dans les cellules de la rétine ne devraient pas pouvoir se déplacer vers d’autres parties du corps. Et comme l’œil est un site immunoprivilégié, le système immunitaire a moins de chances d’y organiser une défense contre un tel virus.
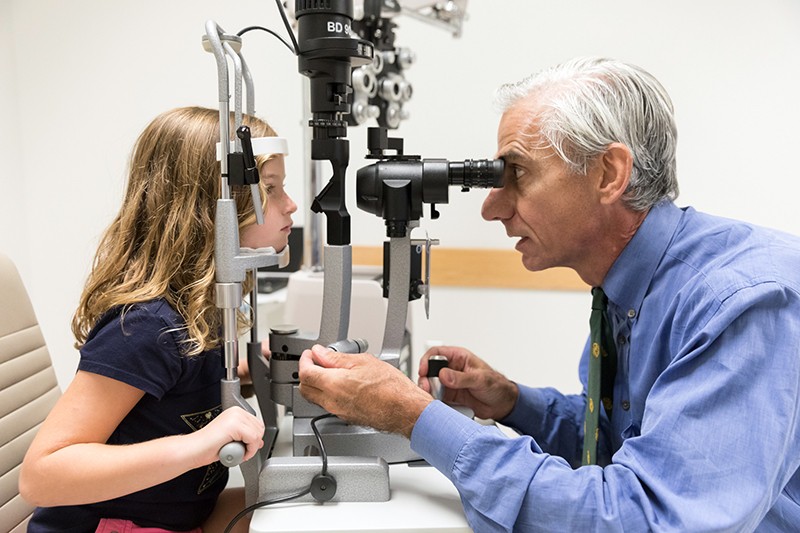
L’ophtalmologue Albert Maguire examine les yeux d’une fillette atteinte d’amaurose congénitale de Leber dont la vue a été restaurée par la thérapie génique voretigene neparvovec (Luxturna).Crédit : Hôpital pour enfants de Philadelphie
Dans la première démonstration du potentiel de la thérapie génique pour lutter contre la cécité, trois équipes de chercheurs ont utilisé cette technique pour traiter avec succès des personnes atteintes d’amaurose congénitale de Leber (ACL). Cette maladie héréditaire entraîne de graves déficiences visuelles et commence dès les premières années de la vie. Elle se manifeste souvent par une cécité nocturne avant d’évoluer vers une perte de vision large qui commence à la périphérie du champ visuel. Elle touche environ 1 bébé sur 40 000.
Les chercheurs ont entrepris de s’attaquer à une forme spécifique de l’affection connue sous le nom d’ACL 2. Celle-ci est causée par des mutations dans le RPE65, un gène qui est exprimé par l’EPR. Le gène muté affecte la fonction de l’EPR, ce qui endommage les cellules photoréceptrices. En 2008, les trois équipes, dont l’une dirigée par Hauswirth, ont chacune montré, lors d’essais cliniques précoces, que l’administration d’une copie saine du RPE65 dans la rétine était sûre et entraînait des améliorations limitées de la vision4,5,6. Un essai clinique de phase III dirigé par Albert Maguire, ophtalmologiste à l’Université de Pennsylvanie à Philadelphie, a montré en août 2017 que les personnes atteintes d’ACL 2 ayant reçu le traitement étaient mieux à même de parcourir des courses d’obstacles à différents niveaux d’éclairage que celles qui ne l’avaient pas reçu7. En décembre 2017, la FDA a approuvé le traitement, voretigene neparvovec (Luxturna), ce qui en fait la première thérapie génique, toutes pathologies confondues, à obtenir le feu vert pour une utilisation clinique.
Il est possible de traiter l’ACL 2 de cette manière car les mutations génétiques en cause présentent un mode d’hérédité récessif. Cela signifie que les deux copies de RPE65 d’une personne doivent porter les mutations pertinentes pour provoquer le trouble. L’apport d’une seule version non mutée résout donc le problème. Les maladies causées par des mutations à transmission dominante, en revanche, ne nécessitent qu’une seule copie mutée d’un gène pour se manifester. Dans la plupart de ces cas, le simple ajout d’une copie normale du gène n’est d’aucune utilité ; le gène muté doit être inactivé. Une option consiste à le réduire au silence en ajoutant des molécules d’ARN spécifiques qui interceptent les instructions du gène muté pour la fabrication de la protéine défectueuse, puis en fournissant une copie normale du gène pour qu’il reprenne ses fonctions – une approche appelée suppression et remplacement. Une autre méthode consiste à corriger la mutation à l’aide de la technique d’édition de gènes CRISPR-Cas9. Des chercheurs de l’université de Modène et Reggio Emilia à Modène, en Italie, ont démontré cette approche dans un modèle de souris de rétinite pigmentaire8 en 2016. L’année suivante, une équipe américaine l’a utilisée pour corriger la mutation à l’origine d’un type de glaucome à la fois chez la souris et dans des cellules humaines en culture9.
Un moteur important des progrès de la thérapie génique a été l’utilisation du virus adéno-associé (AAV) pour délivrer des gènes de remplacement aux cellules. Les AAV se sont révélés sûrs, en partie parce qu’ils ont tendance à ne pas s’intégrer dans le génome de leur cellule hôte, ce qui minimise le risque que les cellules deviennent cancéreuses. Et leur petite taille leur permet de se diffuser largement dans l’œil et donc d’infecter un grand nombre de cellules. Mais la capacité des AAV à délivrer des gènes a des limites : certains gènes sont tout simplement trop grands pour être transportés par les AAV, notamment ABCA4, dont les mutations peuvent entraîner la maladie de Stargardt, une forme héréditaire de dégénérescence maculaire. Deux solutions de contournement sont à l’étude. La première consiste à utiliser un virus ayant une plus grande capacité de transport, tel qu’un lentivirus, pour délivrer les gènes de remplacement. La sécurité et l’efficacité de cette approche sont inconnues, mais des essais cliniques sont en cours. Une deuxième stratégie consiste à casser le gène de remplacement en deux et à transporter chaque moitié séparément dans la cellule, ainsi qu’un moyen de les recombiner. « Cela fonctionne dans au moins un modèle animal à l’heure actuelle », déclare Hauswirth.
Quelle que soit l’approche, la thérapie génique présente une limite considérable. Plus de 250 gènes sont impliqués dans la cécité, et comme chacun peut être affecté par de nombreux types de mutation, le nombre de cibles thérapeutiques potentielles est énorme. Par exemple, plus de 100 mutations dans le gène RHO conduisent à la rétinite pigmentaire, la maladie rétinienne à transmission dominante la plus courante. Le développement d’une thérapie génique pour chaque mutation n’est pas pratique, dit Hauswirth.
Les chercheurs travaillent sur une solution potentielle qui apporte une touche à l’approche de suppression et de remplacement. Au lieu de cibler les copies de RHO contenant une mutation spécifique, ils utilisent un ARN silencieux pour supprimer toute expression du gène, que RHO soit muté ou non, tout en délivrant une copie de remplacement qui est immunisée contre l’ARN silencieux. Une équipe dirigée par Jane Farrar, généticienne au Trinity College de Dublin, a montré les promesses de cette stratégie en 2011 dans un modèle de souris de rétinite pigmentaire dominante10. En 2018, Hauswirth et ses collègues ont testé cette approche chez des chiens atteints de rétinite pigmentaire11. Ils ont montré que la dégénérescence des cellules photoréceptrices dans les zones traitées de la rétine pouvait être stoppée – une amélioration qui a persisté pendant au moins huit mois. Cette stratégie s’attaque à toutes les mutations susceptibles de provoquer une rétinite pigmentaire à transmission dominante en un seul traitement, et étend donc la thérapie génique des maladies récessives aux maladies à transmission dominante « d’une manière assez simple », explique Hauswirth. Il prévoit d’étudier dans quelle mesure les chiens qui ont reçu le traitement peuvent naviguer dans un labyrinthe, et recueille les données de sécurité nécessaires au lancement d’un essai clinique.
Optogénétique
La thérapie génique ne fonctionne que chez les personnes dont la cécité est due à une mutation génétique. Elle n’est pas non plus appropriée pour s’attaquer aux maladies rétiniennes en phase terminale, dans lesquelles il reste un nombre insuffisant de cellules à réparer. Mais une approche connexe, basée sur une technique appelée optogénétique, ne tient pas compte des troubles et pourrait conduire à des traitements pour différents stades de dégénérescence. Dans l’optogénétique, les gènes qui permettent aux cellules de produire des protéines sensibles à la lumière, appelées opsines, sont délivrés par un virus. L’introduction d’opsines peut restaurer une certaine sensibilité à la lumière des photorécepteurs endommagés, ou même rendre sensibles à la lumière d’autres cellules de la rétine, notamment les cellules bipolaires ou les cellules ganglionnaires rétiniennes.

L’optogénétique a été utilisée pour restaurer la sensibilité à la lumière des cellules coniques (vertes) dans un modèle de souris de rétinite pigmentaire ; le succès de la technique a été évalué en mesurant l’activité d’une cellule ganglionnaire de la rétine (magenta), qui est stimulée par les cônes en réponse à la lumière.Crédit : IOB.ch
Problématiquement, cependant, alors que les cellules photoréceptrices de l’œil peuvent faire face à une large gamme d’intensités lumineuses – fonctionnant bien à la fois en plein soleil et au crépuscule – les opsines ont une gamme limitée et sont souvent plus performantes à des intensités lumineuses élevées. Une solution potentielle consiste à utiliser un dispositif similaire au système d’œil bionique Prima de Pixium Vision, qui consiste à équiper les patients de lunettes intégrant une caméra vidéo qui capture la vue de l’utilisateur et un projecteur qui pointe vers l’œil. Comme pour Prima, l’avantage est que la nature de la lumière qui entre dans l’œil peut être adaptée à la modification de la rétine ; cependant, dans ce cas, l’intensité et la longueur d’onde choisies sont celles qui pilotent le mieux les opsines nouvellement introduites plutôt que les photodiodes implantées.
GenSight Biologics, une société de biotechnologie de Paris qui compte Sahel et Roska parmi ses fondateurs, teste déjà un tel système. Il vise à délivrer une opsine aux cellules ganglionnaires de la rétine, mais il y a un hic potentiel : les cellules ganglionnaires de la rétine sont naturellement sensibles à la lumière. Elles expriment la mélanopsine, une protéine impliquée dans le réflexe lumineux pupillaire, qui consiste à resserrer la pupille de l’œil en réponse à une lumière vive. Pour éviter de déclencher ce réflexe, les chercheurs de GenSight utilisent une opsine qui répond aux longueurs d’onde de la lumière rouge, car la mélanopsine répond de préférence à la lumière à l’extrémité bleue du spectre. L’entreprise a commencé un essai clinique de stade précoce en octobre 2018 chez des personnes atteintes de rétinite pigmentaire avancée et ayant une vue minimale restante. L’essai impliquera des cohortes du Royaume-Uni, de la France et des États-Unis, et les premiers résultats sont attendus pour la fin de 2020.
« Il s’agit d’une approche simple, et nous devrons voir ce qui sera gagné », déclare Roska. « Ensuite, nous pourrons passer à des approches de plus en plus sophistiquées ». L’un des problèmes qui subsistent est que bon nombre des troubles que les techniques optogénétiques pourraient traiter impliquent une dégénérescence de parties spécifiques de la rétine, la vision utile étant conservée dans d’autres zones. La lumière qui commande les opsines est visible et pourrait interférer avec la vision naturelle restante. À l’avenir, les opsines qui répondent à la lumière proche de l’infrarouge pourraient permettre aux traitements optogénétiques de fonctionner en tandem avec la vision naturelle résiduelle.
Régénération cellulaire
La thérapie par cellules souches pourrait potentiellement guérir la cécité même aux stades avancés de la maladie. Comme les cellules souches peuvent être incitées à devenir n’importe quel type de cellule, elles pourraient être utilisées pour faire pousser des cellules rétiniennes fraîches à transplanter dans l’œil pour remplacer celles qui ont été perdues. Toutefois, des études menées sur des animaux ont montré que seule une petite proportion des neurones transplantés sont capables de s’intégrer correctement dans le circuit neuronal complexe de la rétine. Cela constitue un obstacle considérable pour les traitements à base de cellules souches qui visent à remplacer les neurones de la rétine.

La structure cellulaire complexe de la rétine comprend des couches de photorécepteurs (vert) et de vaisseaux sanguins et de nerfs (magenta).Crédit : Louise Hughes/SPL
Les cellules qui composent l’épithélium pigmentaire de la rétine, en revanche, se situent en dehors des circuits de la rétine. Les thérapies basées sur les cellules souches sont donc les plus prometteuses pour les affections, telles que la DMLA et la rétinite pigmentaire, qui provoquent la dégénérescence des cellules de l’EPR. « Les photorécepteurs doivent se connecter aux circuits, mais pas l’épithélium pigmentaire rétinien », explique Roska. « C’est là que les gens sont le plus près de faire des progrès ». Dans un premier temps, les chercheurs ont essayé d’injecter dans la rétine des cellules de l’EPR dérivées de cellules souches en suspension, mais trop peu sont restées là où elles étaient nécessaires. Plusieurs équipes pensent désormais qu’une meilleure approche consiste à transplanter les cellules EPR dans l’œil sous la forme d’une feuille préformée qui est ensuite maintenue en position par un échafaudage biocompatible. « L’approche par échafaudage est une énorme amélioration, par rapport à la suspension, pour les cellules EPR », explique Sahel.
En mars 2018, le London Project to Cure Blindness – une collaboration entre l’University College London et le Moorfields Eye Hospital de Londres – a annoncé les résultats d’un essai de phase I dans lequel une feuille de cellules EPR a été implantée dans la rétine de deux personnes atteintes de DMLA humide (une forme rare et grave de DMLA impliquant une croissance anormale et une fuite des vaisseaux sanguins). Les deux receveurs ont bien toléré la procédure et ont été capables de lire 21 à 29 lettres de plus sur une grille de lecture qu’avant le traitement12. Le mois suivant, une équipe dirigée par Humayun a fait état de résultats similaires en phase I chez cinq personnes atteintes de DMLA sèche, la forme la plus courante de l’affection13. Ces premiers résultats sont pleins de promesses. « Cela a suscité beaucoup d’enthousiasme », déclare Humayun. Mais les résultats doivent être confirmés par des essais de phase III sur un plus grand nombre de participants, et Humayun prévient que le traitement pourrait être loin d’être utilisé en clinique pendant de nombreuses années, car aucune thérapie à base de cellules souches pour un trouble de la rétine n’a encore franchi le processus d’approbation.
Une approche connexe, encore aux premiers stades de la recherche fondamentale, pourrait combler l’espoir de remplacer les neurones perdus, ouvrant la voie à des traitements pour une grande variété de maladies oculaires. Chez l’homme, les neurones matures ne se divisent pas et ne peuvent donc pas se régénérer, ce qui rend la restauration de la vision particulièrement difficile. Mais il n’en va pas de même pour tous les animaux. Les reptiles et certains poissons peuvent régénérer les neurones de la rétine, et les oiseaux présentent également une certaine capacité de régénération. Thomas Reh, neuroscientifique à l’université de Washington à Seattle, tente de débloquer cette capacité chez l’homme. Mais plutôt que de transplanter des cellules cultivées en laboratoire, Reh vise à amadouer les cellules qui se trouvent déjà dans la rétine pour qu’elles se différencient en neurones frais.
En 2001, Reh a suggéré que la glie de Müller – des cellules qui fournissent une structure à la rétine et soutiennent sa fonction – est la source de nouveaux neurones qui avaient été observés chez les poissons et les oiseaux14. Avec son équipe, il a alors cherché à savoir si la glie de Müller pouvait être utilisée pour générer de nouveaux neurones chez la souris. En 2015, ils ont modifié des souris pour qu’elles produisent Ascl1, une protéine importante pour la production de neurones chez les poissons, puis ont endommagé la rétine des animaux15. Leur espoir était qu’Ascl1 provoque la transformation de la glie de Müller en neurones.
L’expérience n’a pas réussi à produire de nouveaux neurones chez les souris adultes, mais a réussi chez les jeunes souris. Nikolas Jorstad, biochimiste et doctorant dans l’équipe de Rehs, a proposé que les modifications chimiques apportées à la chromatine (un complexe d’ADN, d’ARN et de protéines) dans le noyau cellulaire au cours du développement puissent bloquer l’accès, dans les cellules matures, aux gènes qui permettent à la glie de Müller de se transformer en neurones. En août 2017, l’équipe de Reh a montré qu’en introduisant une enzyme qui inverse ces modifications, elle pouvait inciter la glie de Müller à se différencier16. « Pour la première fois, nous avons pu régénérer des neurones chez la souris adulte », déclare Reh. « Après toutes ces années, j’étais plutôt ravi ». Bien qu’ils ne soient pas de véritables cellules photoréceptrices et qu’ils ressemblent davantage à des cellules bipolaires, les neurones se sont connectés aux circuits existants et étaient sensibles à la lumière. « J’ai été surpris qu’ils se connectent aussi bien », déclare Reh.
Bien que loin d’être prêts à traiter les troubles rétiniens chez l’homme, ces travaux ont un énorme potentiel. La prochaine étape consistera à répéter les études sur des animaux dont les yeux ressemblent davantage à ceux des humains. L’équipe de Reh travaille déjà avec des cultures de cellules rétiniennes provenant de primates non humains. Les chercheurs doivent également trouver comment diriger le processus de différenciation pour produire des types de cellules spécifiques tels que les bâtonnets et les cônes. « Maintenant que nous avons mis le pied dans la fabrication de neurones, des cônes seraient parfaits », déclare Reh.
Si elle réussit, cette approche pourrait être largement applicable. « En fin de compte, ce sera la façon dont toutes ces maladies oculaires seront traitées », prédit Reh. « C’est tout simplement logique. Vous n’avez pas à vous soucier du bon déroulement des transplantations. Vos cellules sont là où vous en avez besoin. »
Humayun est également encouragé par ces travaux. « J’encourage tous ceux qui ont une nouvelle bonne idée », dit-il. « C’est très tôt, c’est à haut risque, mais il ne faut jamais dire jamais. C’est ce que j’ai appris. »