- Introduction
- Materiais e Métodos
- Sample Preparation
- MinION Library Preparation and Sequencing
- Análise Genômica
- Resultados
- MinION Sequenciamento de Dados e Montagem de Genomas Virais
- Extensão e Fonte de Contaminação de Amostra Cruzada
- Discussão
- Contribuições dos autores
- Funding
- Conflito de interesses
- Agradecimentos
- Material Suplementar
- Notas de rodapé
Introduction
Sequenciamento Metagenômico tem o potencial de permitir a identificação imparcial de patógenos a partir de uma amostra clínica. Ela tem a promessa de servir como um ensaio único e universal para diagnóstico de doenças infecciosas diretamente de amostras sem a necessidade de conhecimento a priori (Bibby, 2013; Miller et al., 2013; Schlaberg et al., 2017). Além da identificação de espécies patogénicas, dados de sequência metagenómica ampla e profunda podem fornecer informações relevantes para a determinação do tratamento e prognóstico, detecção de surtos e rastreio da epidemiologia da infecção (Greninger et al., 2010; Yang et al., 2011; Qin et al., 2012; Loman et al., 2013). As plataformas de sequenciamento de próxima geração (NGS) podem produzir uma produção massiva de dados a um custo modesto, porém sua aplicação no diagnóstico clínico e na saúde pública tem sido limitada pela complexidade, lentidão e investimento de capital.
O MinION é um sequenciador de genoma de uma só molécula, em tempo real, desenvolvido pela Oxford Nanopore Technologies (ONT). O tamanho compacto e a natureza em tempo real do MinION poderiam facilitar a aplicação do sequenciamento metagenômico em testes de ponto de tratamento de doenças infecciosas, como demonstrado por vários estudos de prova de conceito, incluindo a identificação de Chikungunya (CHIKV), Ebola (EBOV) e vírus da hepatite C (HCV) a partir de amostras de sangue clínico humano sem enriquecimento do alvo (Greninger et al, 2015), e detecção de patógenos bacterianos a partir de amostras de urina (Schmidt et al., 2016) e amostras respiratórias, sem a necessidade de cultura prévia (Pendleton et al., 2017).
A produção de dados de MinION aumentou muito desde sua liberação em 2015, com cada célula de fluxo consumível gerando agora até 10-20 Gb de dados de seqüência de DNA. Isto permite aos usuários fazer um uso mais eficiente da célula de fluxo (e reduzir o custo) através da multiplexação de várias amostras em uma única execução sequencial. A ONT desenvolveu conjuntos de códigos de barras sem PCR que permitem a multiplexação de até 12 amostras.
Detecção do vírus da gripe A em várias amostras respiratórias pode ser uma utilização de diagnóstico de um ensaio de sequenciação MinION multiplexado. No entanto, ao seqüenciar diretamente de amostras com uma ampla gama de títulos virais em potencial, é importante estar ciente do potencial de contaminação de amostras cruzadas, tanto durante a preparação da biblioteca quanto na fase de desmultiplexação do código de barras bioinformático após o seqüenciamento. Aqui, apresentamos um conjunto de dados exclusivo de sequenciamento MinION e resultados de investigação sobre a extensão e fonte da contaminação por código de barras cruzado no sequenciamento multiplex.
Materiais e Métodos
Utilizamos uma amostra de lavagem nasal de furão infectado com o vírus da gripe A como exemplo e também fizemos um pico em duas alíquotas de amostras negativas de lavagem nasal de furão não infectado (estoques pré-existentes não utilizados de um estudo não relacionado) com vírus de dengue e chikungunya separadamente. Nenhum destes vírus é relevante para o diagnóstico clínico em amostras respiratórias, mas agem aqui como marcadores claros e distintos para a avaliação da contaminação de amostras cruzadas. As bibliotecas de sequenciação para cada amostra foram preparadas em paralelo, juntamente com um controle negativo de lavagem nasal, código de barras e sequenciadas individualmente. Em seguida, reunimos uma alíquota das bibliotecas de seqüenciamento e realizamos o sequenciamento Multiplex MinION. As leituras das quatro séries individuais (referidas como “CHIKV,” “DENV,” “FLU-A,” e “Negative”) e a série multiplexada (referida como “Multiplexed”) foram então analisadas para investigar a extensão e a fonte da contaminação da amostra cruzada.
Sample Preparation
A licença do projeto foi analisada pelo AWERB (Animal Welfare and Ethics Review Board) local e foi posteriormente concedida pelo Home Office. O RNA foi extraído, usando o QIAamp viral RNA kit (Qiagen) de acordo com as instruções do fabricante, a partir de um banho nasal de furão contendo o vírus da gripe A (H1N1) (A/Califórnia/04/2009) e uma piscina de amostras negativas de banho nasal. Alíquotas de extrato de amostra negativa foram picar com dengue (DENV) (cepa TC861HA, GenBank: MF576311) ou CHIKV (cepa S27, GenBank: MF580946.1) RNA viral de The National collection of Pathogenic Viruses1. As amostras foram tratadas com DNase utilizando TURBO DNase (Thermo Fisher Scientific, Waltham, MA, Estados Unidos) e purificadas utilizando o RNA Clean & Kit ConcentratorTM-5 (Zymo Research). cDNA foi preparado e amplificado utilizando um método Sequence-Independent-Single-Primer-Amplification (Greninger et al., 2015) modificado conforme descrito anteriormente (Atkinson et al., 2016). O cDNA amplificado foi quantificado usando o Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA, Estados Unidos), e 1 μg foi usado como entrada para cada preparação da biblioteca MinION, com exceção do controle negativo onde a amostra inteira (32 ng) foi usada.
MinION Library Preparation and Sequencing
Ligation Sequencing Kit 1D (SQK-LSK108) e Native Barcoding Kit 1D (EXP-NBD103) foram utilizados de acordo com os protocolos padrão ONT, com a exceção de que apenas um código de barras foi incluído em cada uma das quatro preparações da biblioteca. Cada biblioteca foi executada em uma célula de fluxo individual e uma quinta biblioteca conjunta foi feita através da combinação das quatro bibliotecas de código de barras individuais. As bibliotecas foram sequenciadas em células de fluxo R9.4. O desenho do estudo é mostrado na Figura 1.
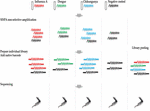
FIGURA 1. Visão geral do desenho do estudo. O RNA foi extraído de quatro amostras, incluindo uma amostra de lavagem nasal de furão infectado com o vírus influenza A, duas amostras negativas de lavagem nasal de furão com picos de dengue e vírus chikungunya, e um controle negativo de lavagem nasal de furão. cDNA foi preparado e amplificado usando um método de amplificação Sequence-Independent-Single-Primer-Amplification. As bibliotecas de sequenciamento para cada amostra foram preparadas em paralelo, em código de barras, e sequenciadas em células de fluxo individuais. O sequenciamento multiplexado também foi realizado através do agrupamento das quatro bibliotecas individuais. As leituras das quatro execuções individuais e da execução multiplex foram analisadas para avaliar a extensão e fonte da contaminação por código de barras cruzado no sequenciamento multiplex.
Análise Genômica
Leituras foram chamadas de base usando Albacore v2.1.7 (ONT) com demultiplexação do código de barras. As leituras de cada sequência foram mapeadas para sequências genómicas de cada vírus usando Minimap2 (Li, 2018). O número de leituras mapeadas para referência foi contado usando o Pysam2. A montagem de novo foi realizada usando Canu v1.7 (Koren et al., 2017), e o esboço resultante do genoma foi polido usando Nanopolish (Mongan et al., 2015) com os dados em nível de sinal.
Para permitir a demultiplexação rigorosa dos dados de sequenciação de Minion Multiplex, fizemos duas rodadas de análises usando Porechop (v0.2.23). A presença de sequência de adaptação no meio de uma leitura é uma assinatura de quimera. Usamos Porechop para examinar cada leitura e aqueles que têm partilha da região média >75% de identidade com sequência de adaptação foram identificados como leituras quiméricas. Em Porechop, definimos a opção “-middle_threshold” e escolhemos um limiar de 75. Na segunda ronda, utilizámos Porechop para procurar a sequência de códigos de barras tanto no início como no final de uma leitura; as leituras foram atribuídas apenas se o mesmo código de barras fosse encontrado em duas extremidades. Definimos a opção “-require_two_barcodes” no Porechop e definimos o limite para a pontuação do código de barras como 70. Para encontrar a assinatura potencial de leituras quiméricas, examinamos os sinais atuais de leitura armazenados no arquivo FAST5 pelo seqüenciador MinION. Os sinais correntes foram extraídos usando ONT fast5 API4 e plotados usando ggplot2 implementado em R5 para uma comparação de leituras quiméricas e não quiméricas.
Resultados
MinION Sequenciamento de Dados e Montagem de Genomas Virais
O rendimento de cada execução de sequenciamento MinION variou devido a diferenças no tempo de execução. Um número máximo de leituras de ∼2.4 M foi alcançado pela execução do sequenciamento multiplexado e a execução individual de CHIKV, devido ao maior tempo de execução (Tabela Suplementar S1). As leituras do vírus spiked representaram 96% dos dados na execução individual do CHIKV e do DENV, e 78% para a amostra FLU-A (Tabela 1). A percentagem de leituras virais dentro de cada amostra com código de barras nos dados do sequenciamento Multiplexed está próxima da percentagem nos dados da amostra executada individualmente (Tabela 2). Cada genoma viral tinha um ultra-alto (>8.000) profundidade média de cobertura nos dados do sequenciamento individual e multiplexado, e o conjunto de novo foi capaz de recuperar genomas quase completos para todos os três vírus com 99.9% identidades comparadas com a referência do GenBank.

TABLE 1. Resumo dos resultados do mapeamento e da montagem de novo para dados do sequenciamento MinION de bibliotecas individuais.

TABLE 2. Resumo dos resultados do mapeamento e da montagem de novo para dados de multiplexação MinION.
Extensão e Fonte de Contaminação de Amostra Cruzada
Cada amostra foi codificada com código de barras, e sequenciada individualmente e multiplexada, o que nos permitiu examinar o desempenho da demultiplexação de código de barras do Albacore. Nos dados da amostra sequenciada individualmente, esperaríamos apenas a presença de um único código de barras nativo. Para as sequências individuais de CHIKV (código de barras NB01), DENV (NB09) e FLU-A (NB10), constatamos que 86, 109 e 17 leituras, respectivamente, foram atribuídas a caixas com código de barras não esperadas na biblioteca (representando 0,0036, 0,0129 e 0,001% do total de leituras). Nos dados do sequenciamento multiplex, 41 leituras (0,0016%) foram atribuídas a códigos de barras não incluídos nos experimentos (ou seja, um código de barras diferente de NB01, NB05, NB09, ou NB10). Definimo-las como leituras mal atribuídas (Figura 2A).
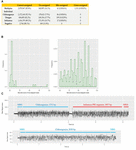
FIGURA 2. (A) resumo do número e porcentagem de leituras corretamente atribuídas, não atribuídas, mal atribuídas e cruzadas em cada execução de sequenciamento. Un-assigned refere-se a leituras que não podem ser atribuídas a nenhuma caixa pelo Albacore devido a uma pontuação de código de barras inferior a 60, mis-assigned refere-se a leituras que foram atribuídas a caixas com código de barras não incluídas neste experimento, e cross-assigned refere-se a leituras que foram atribuídas às caixas com código de barras incorreto; (B) distribuição das pontuações de código de barras relatadas pelo Albacore para leituras mal atribuídas e leituras cruzadas nos dados do sequenciamento multiplex; (C) comparação do sinal bruto de um quimérico e uma leitura corretamente atribuída. O sinal de leitura quimérica possui um sinal de parada e um enorme sinal de pico no meio da leitura.
Para examinar a contaminação potencial do laboratório na preparação da biblioteca de sequenciação, mapeamos todas as leituras de cada execução individual contra as sequências genómicas dos três vírus. Não foi encontrada nenhuma leitura originada de um genoma preparado em uma biblioteca diferente, sugerindo a ausência de contaminação in vitro. A biblioteca de sequenciamento multiplex foi preparada reunindo as bibliotecas individuais não contaminadas após a ligação do código de barras e do adaptador. Entretanto, os resultados do mapeamento mostram 1.311 (0,0543%) leituras mapeadas para o genoma alvo incorreto, implicando que elas foram atribuídas cruzadas às caixas de código de barras erradas (mais tarde referidas como “leituras atribuídas cruzadas”), apesar do fato de que a biblioteca de sequenciamento multiplexado foi agrupada com bibliotecas individuais que não mostraram nenhuma leitura atribuída cruzada. Nós colocamos a hipótese de que as leituras mal atribuídas e cruzadas se deviam a uma pontuação baixa do código de barras, e investigamos as pontuações dos códigos de barras dessas leituras. A maioria das leituras mal atribuídas tinha uma pontuação de código de barras <70, no entanto, as leituras mal atribuídas tinham pontuações mais diversas, variando de 60 a quase 100 (Figura 2B). Este resultado sugeriu que as leituras mal atribuídas e cruzadas têm origem em fontes diferentes. Nós fizemos uma análise das leituras cruzadas em uma pequena base de dados que compreende as seqüências genômicas dos três vírus incluídos neste estudo, e demonstramos que 1074/1311 (82%) destas leituras poderiam ser cruzadas alinhadas a mais de um genoma viral (1.047 leituras) ou cruzadas a regiões distintas dentro do mesmo genoma (27 leituras), sugerindo que são quimeras. Para confirmar esta observação, investigamos os sinais de corrente bruta de algumas leituras cruzadas em comparação com as leituras corretamente atribuídas (Figura 2C). Os sinais de corrente de uma leitura corretamente atribuída geralmente incluem: (i) um sinal de poro aberto de corrente elevada representando o tempo que o poro sequencial muda de um adaptador para outro, (ii) um sinal de paragem, referindo-se ao período de tempo em que uma sequência de ADN está no poro mas ainda não se move, e (iii) o traço de sinal da sequência de ADN. Em contraste, uma leitura quimérica possui um sinal de perda e um enorme sinal de espigão no meio da leitura. As leituras quiméricas podem possuir duas sequências diferentes de códigos de barras no início e no fim, confundindo assim a atribuição de uma caixa de códigos de barras. Em conjunto, estes dados demonstram duas categorias de erro que contribuem para a contaminação cruzada de amostras no nosso conjunto de dados: (i) leituras quiméricas (conta para ∼80% de todas as leituras cruzadas atribuídas); (ii) leituras com pontuação baixa do código de barras. A fim de melhorar a qualidade do nosso conjunto de dados final, exploramos o impacto de diferentes abordagens de demultiplexação de códigos de barras para remover leituras com atribuição cruzada (Tabela 3). A filtragem das leituras que possuem um adaptador interno pode remover 90% das leituras cruzadas e perder 24% do total das leituras. Também tentamos um esquema de filtragem mais rigoroso que requeria dois códigos de barras (um no início e outro no final da leitura) para fazer uma atribuição. Esta abordagem removeu todas as leituras atribuídas, excepto duas, mas perdeu 56% do total de leituras.

TABLE 3. Remoção de leituras cruzadas e perda de dados de sequenciamento total por duas abordagens de filtragem usando Porechop.
Investigamos também a extensão de leituras quiméricas potenciais nos dados de sequenciamento. Para as sequências individuais CHIKV, DENV, e FLU-A, os resultados do mapeamento mostram que 2,3, 3,0, e 2,7% das leituras mapeadas, respectivamente, possuem alinhamento suplementar e alinhadas pelo menos duas vezes ao mesmo genoma (Tabela 4). Consideramos tanto as leituras classificadas e não classificadas por código de barras nos dados do sequenciamento multiplex. Os resultados mostram que 2,0% das leituras mapeadas possuem alinhamento suplementar e alinhadas pelo menos duas vezes ao mesmo genoma, enquanto 0,052% do total de leituras foram alinhadas a pelo menos dois genomas distintos.

TABLE 4. Resumo do número e percentagem de leituras não quiméricas, auto-quiméricas e cruzadas em cada execução de sequenciação.
Discussão
O objectivo final da nossa investigação é desenvolver um ensaio de diagnóstico baseado em sequenciação metagenómica nanopore que permita a realização de testes no local de tratamento de doenças infecciosas. O sequenciamento multiplex oferece a oportunidade de melhorar a escalabilidade e reduzir custos, no entanto, a contaminação cruzada de amostras pode levar a erros nos dados e falsa interpretação dos resultados.
Neste experimento, reunimos bibliotecas limpas e realizamos o sequenciamento multiplex MinION a fim de investigar a extensão e a fonte da contaminação cruzada de códigos. Identificamos que 0,056% do total de leituras foram cruzadas com as caixas de códigos de barras incorretas, o que é comparável ao relatado para as plataformas de seqüenciamento Illumina de diferentes estudos (entre 0,06 e 0,25%) (Nelson et al., 2014; D’Amore et al., 2016; Wright e Vetsigian, 2016). Nossos resultados mostraram que as leituras quiméricas são a fonte predominante de erros de atribuição de códigos cruzados. As leituras quiméricas cruzadas neste conjunto de dados só poderiam ter sido formadas durante o sequenciamento e não durante a preparação da biblioteca, pois estavam completamente ausentes nos dados de sequenciamento de bibliotecas individuais, e a única etapa adicional de processamento era misturar as bibliotecas de sequenciamento final antes do carregamento. Hipótese é que o algoritmo atual implementado no Albacore não consegue reconhecer a curta dissociação entre seqüências de DNA que correm concomitantemente através do nanopore, concatenando assim mais de uma seqüência no mesmo arquivo Fast5.
Leituras quiméricas foram observadas em dados de seqüenciamento MinION antes em White et al. (2017). Através da análise dos dados da seqüência MinION de três diferentes amplicons de interferon, os autores descobriram que 1,7% das leituras mapeadas eram quimeras. Os nossos achados aumentam o conhecimento de que as quimeras são comuns nos dados de sequenciamento de Minion. Identificamos entre 2 e 3% do total de leituras em três indivíduos e um dado de sequenciamento multiplex são quimeras. Nosso estudo difere do trabalho anterior nos dois aspectos seguintes. Primeiro, fornecemos evidências diretas de que leituras quiméricas podem ser formadas após a preparação da biblioteca e durante o seqüenciamento; vinculamos ainda essas quimeras à contaminação por amostras cruzadas no seqüenciamento de MÍNIO Múltiplo, como discutido acima. Por outro lado, nossa configuração experimental tem limitação na identificação de quimeras potenciais formadas na preparação da biblioteca, particularmente durante a etapa de ligação do adaptador no protocolo padrão de seqüenciamento multiplex. Em segundo lugar, nossos resultados refletem o status atual do seqüenciamento MinION porque usamos um novo e mais representativo kit de seqüenciamento ONT, incluindo o kit de seqüenciamento de ligação 1D (SQK-LSK108) e o kit de código de barras nativo 1D (EXP-93 NBD103). A tecnologia de seqüenciamento Nanopore está em rápido desenvolvimento e melhorias estão acontecendo em todos os aspectos. Por exemplo, o novo kit de sequenciamento de ligação de ADN (SQK-LSK109) e o kit de sequenciamento de RNA directo (SQK-RNA001) foram lançados; o algoritmo de basecaller implementado em Albacore e Guppy basecaller foi actualizado. Todas estas alterações têm efeito sobre a extensão da quimera nos dados de sequenciamento de Nanopore e contaminação cruzada de códigos durante o sequenciamento multiplex. A limitação deste estudo foi o pequeno número de experimentos, trabalho adicional usando diferentes configurações de experimentos adicionaria ao nosso entendimento dos dados de seqüenciamento de multiplexação em Nanopore. Além disso, é importante investigar as contribuições de fatores potenciais para a contaminação por código de barras cruzado, o que esclareceria as melhores práticas para analisar dados de sequenciamento de multiplexação.
Em resumo, nosso estudo demonstrou que as leituras quiméricas são a fonte predominante de erros de atribuição de código de barras cruzado no sequenciamento de multiplexação MinION. Ele destaca a necessidade de uma filtragem cuidadosa dos dados de sequenciamento multiplex MinION antes da análise posterior, e o trade-off entre sensibilidade e especificidade que se aplica aos métodos de demultiplexação de código de barras.
Contribuições dos autores
SP, KL, SL, e YX conduziram o sequenciamento MinION. YX analisou os dados. Todos os autores desenharam o estudo, participaram da interpretação dos resultados e escreveram o manuscrito, e leram e aprovaram a versão final deste manuscrito.
Funding
Este trabalho foi apoiado pelo NIHR Oxford Biomedical Research Centre.
Conflito de interesses
Os autores declaram que a pesquisa foi conduzida na ausência de qualquer relação comercial ou financeira que pudesse ser interpretada como um potencial conflito de interesses.
Agradecimentos
Gostaríamos de agradecer ao Dr. Anthony Marriott (Public Health England) por fornecer aspirados nasais de furão.