In tutto il mondo, 36 milioni di persone hanno una perdita totale della vista1. Non riescono a vedere le forme e nemmeno le fonti di luce. Per la maggior parte di queste persone, la loro cecità deriva da problemi rettificabili come la cataratta – semplicemente non hanno accesso a cure sanitarie adeguate. I restanti milioni, tuttavia, sono ciechi a causa di condizioni che attualmente non hanno un trattamento efficace.
“La cecità è una delle condizioni che più alterano la vita di una persona”, dice William Hauswirth, un oftalmologo dell’Università della Florida a Gainesville. Oltre alle difficoltà che provoca per la mobilità e nel trovare lavoro, la menomazione visiva è associata a una serie di altri problemi di salute, tra cui insonnia, ansia e depressione, e anche il rischio di suicidio. “Ripristinare la visione utile sarebbe un miglioramento quasi inimmaginabile della qualità della vita”, dice Hauswirth.
Nei paesi ad alto reddito, dove le cause prevenibili di disabilità visiva sono affrontate di routine, la principale causa di cecità è la degenerazione della retina. Situato nella parte posteriore dell’occhio, questo tessuto contiene cellule specializzate che reagiscono alla luce ed elaborano i segnali visivi, ed è quindi fondamentale per la visione. Le cellule fotorecettrici – neuroni comunemente noti come bastoncelli e coni – convertono la luce che colpisce la retina in segnali elettrochimici. Questi segnali filtrano poi attraverso una complessa rete di altri neuroni, comprese le cellule bipolari, le cellule amacrine e le cellule orizzontali, prima di raggiungere i neuroni noti come cellule gangliari della retina. Le lunghe proiezioni, o assoni, di queste cellule formano il nervo ottico, lungo il quale i segnali dalla retina vengono trasportati alla corteccia visiva del cervello, dove vengono interpretati come immagini.
I disturbi della retina comportano comunemente la perdita di cellule fotorecettrici, che riduce la sensibilità dell’occhio alla luce. In alcuni disturbi retinici, tra cui la degenerazione maculare legata all’età (AMD), questa perdita deriva dal fallimento delle cellule epiteliali che formano uno strato sul retro della retina noto come epitelio pigmentato retinico (RPE). L’RPE mantiene sane le cellule dei fotorecettori ripulendo i sottoprodotti tossici prodotti durante la reazione con la luce, oltre a fornire nutrienti. Nei disturbi retinici in cui i fotorecettori rimangono in buona forma, la principale causa di cecità è la degenerazione delle cellule gangliari retiniche.
Ascolta una versione audio di questo articolo
Il tuo browser non supporta l’elemento audio.
La varietà delle cause della disabilità visiva rende più difficile trovare soluzioni. Ma i progressi in diverse aree stanno aumentando le speranze che quasi tutte le forme di disturbo della retina potrebbero diventare trattabili.
Un approccio è quello di aumentare o bypassare gli occhi danneggiati con protesi funzionali. Tali occhi bionici possono ripristinare solo una visione limitata al momento, ma i ricercatori continuano a spingere le capacità dei dispositivi. Un’altra opzione è la terapia genica. Già disponibile per le persone con specifiche mutazioni genetiche, i ricercatori stanno cercando di estendere questo approccio a più persone e condizioni. Alcuni scienziati stanno anche perseguendo trattamenti basati su una tecnica correlata nota come optogenetica, che comporta l’alterazione genetica delle cellule per ripristinare la sensibilità alla luce della retina. Questo lavoro è in una fase iniziale, ma i ricercatori sperano che l’approccio sarà alla fine in grado di aiutare una vasta gamma di persone, perché è agnostico alle cause della degenerazione retinica. E gli sforzi per sostituire le cellule perse o danneggiate della retina, sia in situ che attraverso trapianti di cellule, suggeriscono che anche i disturbi della retina all’ultimo stadio potrebbero alla fine diventare trattabili.
Molto di questa ricerca è nella sua infanzia. Ma Hauswirth è ottimista sui progressi che sono già stati fatti. Dieci anni fa, dice, spesso doveva dire ai pazienti che non poteva fare nulla per loro. “
Occhi bionici
Quasi 30 anni fa, Mark Humayun, un ingegnere biomedico della University of Southern California di Los Angeles, ha iniziato a stimolare elettricamente la retina delle persone con cecità. Lavorando con i colleghi della Second Sight Medical Products, una società di tecnologia medica a Sylmar, California, i suoi esperimenti hanno dimostrato che tale stimolazione potrebbe indurre la percezione visiva di macchie di luce chiamate fosfeni. Dopo un decennio di lavoro sugli animali per stabilire la quantità di corrente elettrica che poteva essere applicata in modo sicuro all’occhio, e armati di una conoscenza notevolmente aumentata sul numero e sui tipi di cellule che persistono nelle retine umane in degenerazione, il team di Humayun era pronto per iniziare a lavorare con le persone. Tra il 2002 e il 2004, i ricercatori hanno impiantato un occhio bionico in ciascuna delle sei persone che avevano una cecità totale o quasi totale in un occhio – il primo esperimento di questo tipo. I destinatari del dispositivo, noto come Argus I, hanno riferito di essere in grado di percepire fosfeni, movimenti direzionali e persino forme2. Circa 300 persone ora sperimentano il mondo attraverso il successore di quel dispositivo, l’Argus II, che è stato approvato dai regolatori in Europa nel 2011 per l’uso in persone con retinite pigmentosa – un gruppo di rare malattie genetiche che causano la degenerazione delle cellule fotorecettori. La US Food and Drug Administration (FDA) ha seguito l’esempio due anni dopo.

L’impianto Argus II comprende un array di elettrodi che è attaccato alla superficie della retina.Credit: Ringo Chiu/ZUMA /Alamy
Per essere dotati di un Argus II, i pazienti subiscono un intervento chirurgico per attaccare un chip contenente un array di elettrodi alla superficie della retina. Per “vedere” con il dispositivo, una videocamera in miniatura montata su un paio di occhiali trasmette segnali a un’unità di elaborazione che viene indossata dal paziente. Il processore converte i segnali in istruzioni che vengono trasmesse senza fili al dispositivo impiantato. Gli elettrodi stimolano quindi le cellule gangliari della retina nella parte anteriore della retina. L’uso della protesi è un processo di apprendimento. I destinatari devono allenare il loro cervello a interpretare il nuovo tipo di informazioni ricevute. E poiché la videocamera non segue il movimento dell’occhio, devono anche imparare a muovere la testa per dirigere lo sguardo.
Il dispositivo fornisce solo una visione limitata. Gli utenti possono rilevare fonti di luce e oggetti con bordi ad alto contrasto, come porte o finestre, e alcuni possono decifrare lettere grandi. Queste limitazioni sorgono in parte perché i 60 elettrodi del dispositivo forniscono una risoluzione molto bassa rispetto ai milioni di cellule fotorecettrici in un occhio sano. Ma anche questo minimo miglioramento può migliorare notevolmente la vita delle persone.
Mentre l’Argus II è un impianto epiretinico – il che significa che si trova sulla superficie della retina – altri dispositivi in sviluppo sono progettati per essere collocati sotto la retina. Questi impianti subretinici possono stimolare le cellule che sono più vicine a quelle che normalmente introducono segnali alla retina – le cellule fotorecettrici. Stimolando le cellule più in alto nel percorso visivo, i ricercatori sperano di preservare più dell’elaborazione del segnale che viene eseguita da una retina sana.
Retina Implant, una società di biotecnologie con sede a Reutlingen, in Germania, ha costruito un impianto subretinico che comprende fotodiodi (dispositivi semiconduttori che convertono la luce in corrente elettrica) che percepiscono direttamente la luce che entra nell’occhio. Questo elimina la necessità di una videocamera esterna, permettendo agli utenti di dirigere il loro sguardo in modo naturale. L’alimentazione è fornita da un’unità portatile, attraverso una bobina che viene impiantata sotto la pelle sopra l’orecchio. Alpha AMS, la versione attuale del sistema, ha ricevuto l’approvazione normativa in Europa per l’uso in persone con retinite pigmentosa.
Pixium Vision a Parigi sta testando un impianto fotovoltaico subretinico chiamato Prima. Il sistema proietta segnali da una videocamera montata sugli occhiali nell’occhio usando la luce del vicino infrarosso, la cui lunghezza d’onda guida in modo ottimale i fotodiodi nel dispositivo per stimolare le cellule della retina. Proiettare le immagini in questo modo dà agli utenti un certo controllo sulla direzione del loro sguardo, perché possono esplorare la scena muovendo solo i loro occhi. L’alimentazione è fornita anche dalla luce del vicino infrarosso, rendendo l’impianto senza fili e l’intervento chirurgico per montarlo meno complicato. “I pazienti stanno imparando a riacquistare la vista più velocemente, e la risoluzione sembra migliore”, dice José-Alain Sahel, un oftalmologo dell’Università di Pittsburgh, Pennsylvania, che sta conducendo prove di sicurezza del dispositivo in dieci persone con AMD. “
Tutti questi dispositivi funzionano solo quando le cellule funzionanti rimangono nella retina. Nelle comuni condizioni dell’occhio che colpiscono principalmente le cellule dei fotorecettori, tra cui la retinite pigmentosa e la AMD, di solito rimangono alcune cellule da stimolare. Ma quando troppe cellule gangliari retiniche muoiono, come accade nella retinopatia diabetica avanzata e nel glaucoma, tali impianti non possono aiutare. Per le persone senza alcuna funzione retinica residua, a causa di malattie o lesioni, un approccio bionico alternativo potrebbe essere più rilevante.
Humayun e i suoi colleghi stanno lavorando su un sistema che bypassa l’occhio inviando segnali direttamente al cervello. L’idea non è nuova: negli anni ’70, l’ingegnere biomedico statunitense William Dobelle ha dimostrato che stimolando direttamente la corteccia visiva si attivava la percezione dei fosfeni3. Ma la tecnologia degli occhi bionici sta recuperando solo ora. Second Sight ha sviluppato Orion, un sistema che è, secondo Humayun, “fondamentalmente un Argus II modificato”. Come l’originale, utilizza una videocamera e un processore di segnali che comunicano senza fili con un impianto, ma il chip è posto sulla superficie della corteccia visiva piuttosto che sulla retina. Il dispositivo è stato testato su cinque persone con percezione della luce limitata o assente a causa di una lesione oculare o di un danno alla retina o al nervo ottico. “Finora i risultati sono buoni”, dice. “
Dato che una parte della tecnologia è già stata sperimentata nelle persone, Humayun è ottimista sul fatto che il sistema potrebbe ricevere l’approvazione normativa entro pochi anni. “Ovviamente, la chirurgia cerebrale ha un diverso livello di rischio, ma la procedura è abbastanza semplice, e l’Orion potrebbe aiutare molti più pazienti”, dice. Tuttavia, si sa molto meno sulla stimolazione del cervello per fornire una visione utile. “Sappiamo molto sulla retina ma molto poco sulla corteccia”, dice Botond Roska, un neurobiologo dell’Istituto di oftalmologia molecolare e clinica di Basilea in Svizzera. “Ma non ne sapremo mai abbastanza se non ci proviamo”, dice.
Terapia genica
L’occhio è un obiettivo ideale per la terapia genica. Poiché è relativamente autonomo, i virus che sono usati per portare i geni nelle cellule della retina non dovrebbero essere in grado di viaggiare in altre parti del corpo. E poiché l’occhio è un sito immunoprivilegiato, è meno probabile che il sistema immunitario vi organizzi una difesa contro un tale virus.
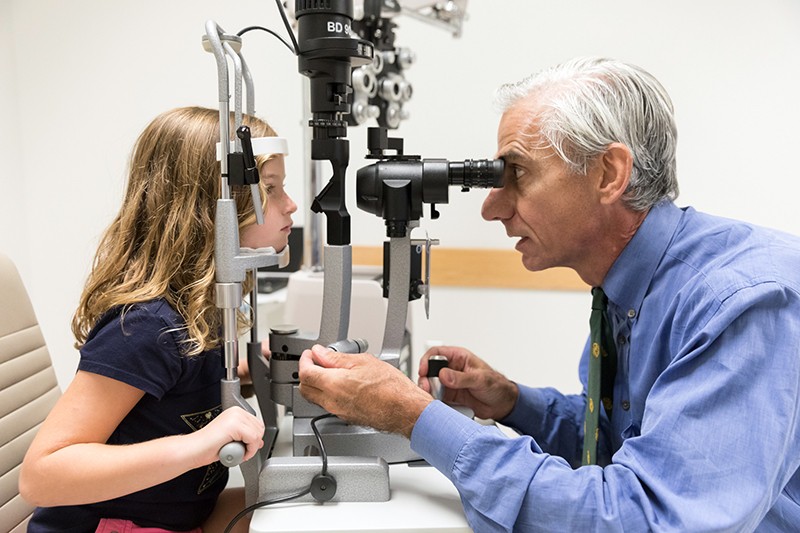
Oftalmologo Albert Maguire esamina gli occhi di una ragazza con amaurosi congenita di Leber la cui vista è stata ripristinata dalla terapia genica voretigene neparvovec (Luxturna).Credit: Children’s Hospital of Philadelphia
Nella prima dimostrazione del potenziale della terapia genica per affrontare la cecità, tre team di ricercatori hanno usato la tecnica per trattare con successo persone con amaurosi congenita di Leber (LCA). Questa condizione ereditaria porta ad una grave menomazione visiva e inizia nei primi anni di vita, spesso manifestandosi come cecità notturna prima di progredire verso un’ampia perdita della vista che inizia alla periferia del campo visivo. Colpisce circa 1 bambino su 40.000.
I ricercatori hanno deciso di affrontare una forma specifica della condizione nota come LCA 2. Questo è causato da mutazioni nel RPE65, un gene che viene espresso dal RPE. Il gene mutato influisce negativamente sulla funzione del RPE, che a sua volta danneggia le cellule fotorecettrici. Nel 2008, i tre team, compreso quello guidato da Hauswirth, hanno dimostrato ciascuno in studi clinici di fase iniziale che la consegna di una copia sana di RPE65 alla retina era sicura e portava a miglioramenti limitati nella visione4,5,6. Uno studio clinico di fase III guidato da Albert Maguire, un oftalmologo dell’Università della Pennsylvania a Filadelfia, ha dimostrato nell’agosto 2017 che le persone con LCA 2 che hanno ricevuto il trattamento erano in grado di navigare meglio nei percorsi a ostacoli a vari livelli di illuminazione rispetto a coloro che non lo hanno fatto7. Nel dicembre 2017, la FDA ha approvato il trattamento, voretigene neparvovec (Luxturna), rendendolo la prima terapia genica per qualsiasi condizione per ottenere la luce verde per uso clinico.
È possibile trattare LCA 2 in questo modo perché le mutazioni genetiche coinvolte mostrano un modello recessivo di eredità. Ciò significa che entrambe le copie di RPE65 di una persona devono portare le mutazioni rilevanti per causare il disturbo. Fornire una singola versione non mutata risolve quindi il problema. Le condizioni che sono causate da mutazioni ereditarie dominanti, tuttavia, richiedono solo una copia mutata di un gene per manifestarsi. Nella maggior parte di queste, la semplice aggiunta di una copia normale del gene non aiuterà; invece, il gene mutato deve essere inattivato. Un’opzione è quella di silenziarlo aggiungendo specifiche molecole di RNA che intercettano le istruzioni del gene mutato per produrre la proteina difettosa, e poi fornendo una copia normale del gene per assumere i suoi compiti – un approccio chiamato soppressione e sostituzione. Un altro è quello di correggere la mutazione usando la tecnica di editing genico CRISPR-Cas9. I ricercatori dell’Università di Modena e Reggio Emilia a Modena, Italia, hanno dimostrato questo approccio in un modello murino di retinite pigmentosa8 nel 2016. L’anno successivo, un team negli Stati Uniti l’ha usato per correggere la mutazione che causa un tipo di glaucoma sia nei topi che nelle cellule umane in coltura9.
Un importante motore del progresso della terapia genica è stato l’uso del virus adeno-associato (AAV) per fornire geni sostitutivi alle cellule. Gli AAV hanno dimostrato di essere sicuri, in parte, perché tendono a non integrarsi nel genoma della cellula ospite, il che minimizza il rischio che le cellule diventino cancerose. E le loro piccole dimensioni permettono loro di diffondersi ampiamente attraverso l’occhio e quindi di infettare un gran numero di cellule. Ma la capacità degli AAV di trasmettere i geni ha dei limiti: alcuni geni sono semplicemente troppo grandi per essere trasportati dagli AAV, tra cui ABCA4, le cui mutazioni possono portare alla malattia di Stargardt, una forma ereditaria di degenerazione maculare. Si stanno cercando due soluzioni. Il primo utilizza un virus con una maggiore capacità di trasporto, come un lentivirus, per consegnare i geni di sostituzione. La sicurezza e l’efficacia di questo approccio non sono note, ma sono in corso studi clinici. Una seconda strategia è quella di spezzare il gene sostitutivo in due e trasportare ogni metà separatamente nella cellula, insieme a un mezzo per ricombinarli. “Questo sta funzionando in almeno un modello animale in questo momento”, dice Hauswirth.
A prescindere dall’approccio, la terapia genica ha un notevole limite. Più di 250 geni sono implicati nella cecità, e poiché ognuno può essere colpito da numerosi tipi di mutazione, il numero di potenziali obiettivi terapeutici è enorme. Per esempio, più di 100 mutazioni nel gene RHO portano alla retinite pigmentosa, il più comune disordine retinico ereditato in modo dominante. Sviluppare una terapia genica per ogni mutazione non è pratico, dice Hauswirth.
I ricercatori stanno lavorando su una soluzione potenziale che mette una svolta nell’approccio di soppressione e sostituzione. Invece di mirare alle copie di RHO che contengono una mutazione specifica, usano un RNA di silenziamento per sopprimere tutta l’espressione del gene, se RHO è mutato o no, mentre forniscono una copia sostitutiva che è immune all’RNA di silenziamento. Un team guidato da Jane Farrar, una genetista del Trinity College di Dublino, ha mostrato la promessa di questa strategia nel 2011 in un modello murino di retinite pigmentosa dominante10. Nel 2018, Hauswirth e colleghi hanno testato l’approccio nei cani con retinite pigmentosa11. Hanno dimostrato che la degenerazione delle cellule dei fotorecettori nelle aree trattate della retina potrebbe essere arrestata – un miglioramento che ha persistito per almeno otto mesi. Questa strategia affronta tutte le mutazioni che possono causare retinite pigmentosa ereditaria dominante in un unico trattamento, e quindi estende la terapia genica da condizioni ereditarie recessive a dominanti “in un modo abbastanza semplice”, dice Hauswirth. Hauswirth prevede di studiare quanto bene i cani che hanno ricevuto il trattamento possono navigare in un labirinto, e sta raccogliendo i dati di sicurezza necessari per iniziare una sperimentazione clinica.
Optogenetica
La terapia genica funziona solo nelle persone la cui cecità è causata da una mutazione genetica. Inoltre non è appropriata per affrontare la malattia retinica allo stadio finale, in cui rimane un numero insufficiente di cellule da riparare. Ma un approccio correlato basato su una tecnica chiamata optogenetica è agnostico e potrebbe portare a trattamenti per diversi stadi di degenerazione. Nell’optogenetica, i geni che permettono alle cellule di produrre proteine sensibili alla luce note come opsine sono consegnati da un virus. L’introduzione di opsine può ripristinare una certa sensibilità alla luce dei fotorecettori danneggiati, o anche rendere sensibili alla luce altre cellule della retina, comprese le cellule bipolari o le cellule gangliari della retina.

L’aptogenetica è stata usata per ripristinare la sensibilità alla luce delle cellule coniche (verdi) in un modello murino di retinite pigmentosa; il successo della tecnica è stato valutato misurando l’attività di una cellula gangliare della retina (magenta), che è stimolata dai coni in risposta alla luce.Credit: IOB.ch
Problematicamente, però, mentre le cellule fotorecettrici dell’occhio possono far fronte a una vasta gamma di intensità della luce – lavorando bene sia in piena luce solare che al crepuscolo – le opsine hanno una gamma limitata e spesso funzionano meglio ad alte intensità di luce. Una soluzione potenziale è quella di utilizzare un set-up che funziona in modo simile al sistema Prima bionic-eye di Pixium Vision, in cui i destinatari sono dotati di occhiali che incorporano una videocamera che cattura la vista dell’utente e un proiettore che punta nel loro occhio. Come con Prima, il vantaggio è che la natura della luce che entra nell’occhio può essere adattata alla modifica della retina; tuttavia, in questo caso, l’intensità e la lunghezza d’onda scelte sono quelle che meglio guidano le opsine appena introdotte piuttosto che i fotodiodi impiantati.
GenSight Biologics, una società di biotecnologie a Parigi che conta Sahel e Roska tra i suoi fondatori, sta già testando un tale sistema. Mira a fornire un’opsina alle cellule gangliari della retina, ma c’è un potenziale intoppo: le cellule gangliari della retina sono naturalmente sensibili alla luce. Esprimono la melanopsina, una proteina coinvolta nel riflesso della luce pupillare, in cui la pupilla dell’occhio si restringe in risposta alla luce intensa. Per evitare di innescare questo, i ricercatori di GenSight stanno usando un’opsina che risponde alle lunghezze d’onda rosse della luce, perché la melanopsina risponde preferenzialmente alla luce all’estremità blu dello spettro. L’azienda ha iniziato una sperimentazione clinica in fase iniziale nell’ottobre 2018 in persone con retinite pigmentosa avanzata che hanno una vista minima residua. La sperimentazione coinvolgerà coorti del Regno Unito, della Francia e degli Stati Uniti, e i primi risultati sono attesi entro la fine del 2020.
“Questo è un approccio semplice, e dovremo vedere cosa si guadagnerà”, dice Roska. “Poi, potremo passare ad approcci sempre più sofisticati”. Un problema che rimane è che molti dei disturbi che le tecniche optogenetiche potrebbero trattare coinvolgono la degenerazione di parti specifiche della retina, con la visione utile conservata in altre aree. La luce che guida le opsine è visibile e potrebbe interferire con la visione naturale rimanente. In futuro, le opsine che rispondono alla luce nel vicino infrarosso potrebbero consentire ai trattamenti optogenetici di lavorare in tandem con la visione naturale residua.
Rigenerazione cellulare
La terapia con cellule staminali potrebbe potenzialmente curare la cecità anche nelle ultime fasi della malattia. Poiché le cellule staminali possono essere indotte a diventare qualsiasi tipo di cellula, potrebbero essere usate per far crescere cellule retiniche fresche da trapiantare nell’occhio per sostituire quelle che sono state perse. Tuttavia, gli studi sugli animali hanno dimostrato che solo una piccola parte dei neuroni trapiantati sono in grado di integrarsi correttamente nel complesso circuito neurale della retina. Questo è un notevole ostacolo per i trattamenti con cellule staminali che mirano a sostituire i neuroni della retina.

La complessa struttura cellulare della retina comprende strati di fotorecettori (verde) e vasi sanguigni e nervi (magenta).Credit: Louise Hughes/SPL
Le cellule che compongono l’epitelio pigmentato retinico, invece, si trovano al di fuori del circuito della retina. Le terapie basate sulle cellule staminali sono quindi più promettenti per le condizioni, come AMD e retinite pigmentosa, che causano la degenerazione delle cellule RPE. “I fotorecettori devono connettersi al circuito, ma l’epitelio pigmentato retinico no”, dice Roska. “È lì che le persone sono più vicine a fare progressi”. Inizialmente, i ricercatori hanno provato a iniettare la retina con cellule RPE derivate da cellule staminali in sospensione, ma troppo poche sono rimaste dove erano necessarie. Diversi team ora pensano che un approccio migliore sia quello di trapiantare le cellule RPE nell’occhio come un foglio preformato che viene poi tenuto in posizione da un’impalcatura biocompatibile. “L’approccio dell’impalcatura è un enorme miglioramento, rispetto alla sospensione, per le cellule RPE”, dice Sahel.
Nel marzo 2018, il London Project to Cure Blindness – una collaborazione tra l’University College London e il Moorfields Eye Hospital di Londra – ha annunciato i risultati di una sperimentazione di fase I in cui un foglio di cellule RPE è stato impiantato nella retina di due persone con AMD umida (una rara e grave forma di AMD che comporta una crescita anormale e perdite di vasi sanguigni). Entrambi i destinatari hanno tollerato bene la procedura e sono stati in grado di leggere 21-29 lettere in più su una tabella di lettura rispetto a prima del trattamento12. Il mese successivo, un team guidato da Humayun ha riportato risultati simili di fase I in cinque persone con AMD secca, la forma più comune della condizione13. Questi risultati iniziali sono pieni di promesse. “Questo ha portato a molto entusiasmo”, dice Humayun. Ma i risultati devono essere confermati da studi di fase III in un maggior numero di partecipanti, e Humayun avverte che il trattamento potrebbe essere lontano molti anni dall’uso in clinica, perché nessuna terapia a base di cellule staminali per un disturbo della retina ha ancora superato il processo di approvazione.
Un approccio correlato, ancora nelle prime fasi della ricerca di base, potrebbe realizzare la speranza di sostituire i neuroni persi, aprendo la porta a trattamenti per un’ampia varietà di malattie degli occhi. Negli esseri umani, i neuroni maturi non si dividono e quindi non possono rigenerarsi, il che rende il ripristino della visione particolarmente difficile. Ma lo stesso non vale per tutti gli animali. I rettili e alcuni pesci possono rigenerare i neuroni della retina, e anche gli uccelli mostrano una certa capacità rigenerativa. Thomas Reh, un neuroscienziato dell’Università di Washington a Seattle, sta cercando di sbloccare questa capacità negli esseri umani. Ma piuttosto che trapiantare cellule coltivate in laboratorio, Reh mira a convincere le cellule che sono già nella retina a differenziarsi in neuroni freschi.
Nel 2001, Reh ha suggerito che la glia di Müller – cellule che forniscono struttura alla retina e sostengono la sua funzione – sono la fonte di nuovi neuroni che erano stati osservati nei pesci e negli uccelli14. Lui e il suo team hanno quindi deciso di scoprire se la glia di Müller poteva essere usata per generare nuovi neuroni nei topi. Nel 2015, hanno ingegnerizzato i topi per fare Ascl1, una proteina che è importante per la produzione di neuroni nei pesci, e poi hanno danneggiato la retina degli animali15. La loro speranza era che Ascl1 avrebbe provocato la trasformazione della glia di Müller in neuroni.
L’esperimento non è riuscito a produrre nuovi neuroni nei topi adulti, ma è riuscito nei topi giovani. Nikolas Jorstad, un biochimico e studente di dottorato nel team di Rehs, ha proposto che le modifiche chimiche apportate alla cromatina (un complesso di DNA, RNA e proteine) nel nucleo della cellula durante lo sviluppo potrebbero bloccare l’accesso nelle cellule mature ai geni che consentono alla glia Müller di trasformarsi in neuroni. Nell’agosto 2017, il team di Reh ha dimostrato che introducendo un enzima che inverte tali modifiche, potrebbero convincere la Müller glia a differenziarsi16. “Per la prima volta, abbiamo potuto rigenerare i neuroni nel topo adulto”, dice Reh. “Dopo tutti questi anni ero piuttosto entusiasta”. Anche se non erano vere cellule fotorecettrici, e assomigliavano più a cellule bipolari, i neuroni si collegavano al circuito esistente, ed erano sensibili alla luce. “Ero sorpreso che si collegassero così bene come fanno”, dice Reh.
Anche se lontano dall’essere pronto a trattare i disturbi della retina nelle persone, il lavoro ha un enorme potenziale. Il prossimo passo sarà quello di ripetere gli studi in animali con occhi più simili a quelli umani. Il team di Reh sta già lavorando con colture di cellule retiniche di primati non umani. I ricercatori devono anche capire come dirigere il processo di differenziazione per produrre tipi di cellule specifiche come bastoncelli e coni. “Ora abbiamo il piede nel business dei neuroni, i coni sarebbero fantastici”, dice Reh.
Se ha successo, l’approccio potrebbe essere ampiamente applicabile. “In definitiva, questo sarà il modo in cui tutte queste malattie dell’occhio saranno trattate”, prevede Reh. “Ha semplicemente senso. Non devi preoccuparti di fare bene i trapianti. Le tue cellule sono proprio dove ne hai bisogno.”
Humayun è anche incoraggiato dal lavoro. “Faccio il tifo per chiunque abbia una nuova buona idea”, dice. “È molto presto, è ad alto rischio, ma mai dire mai. Questo è quello che ho imparato.”