INLEIDING
Interstitiële longaandoeningen vormen een uiterst heterogene groep aandoeningen die het bestaan van een ontstekingscomponent in het longparenchym gemeen hebben die uiteindelijk tot fibrose kan evolueren en die, vanuit functioneel oogpunt, een restrictieve verandering veroorzaakt die uiteindelijk tot ademhalingsinsufficiëntie kan leiden.
Hoewel het hier gaat om ziekten waarvan de diagnose en de behandeling in de gespecialiseerde pneumologie worden uitgevoerd, wordt de behandeling in wezen poliklinisch gevolgd, en aangezien deze vaak gepaard kan gaan met verschillende complicaties, is het van belang dat de eerstelijnsarts op de hoogte is van de huidige richtsnoeren voor de behandeling van deze ziekten, aangezien hij of zij vaak zal worden geraadpleegd door patiënten met dit soort problemen, hetzij ten gevolge van complicaties van de luchtwegen, hetzij ten gevolge van andere soorten complicaties.
Het doel van dit overzicht is de therapeutische modaliteiten te actualiseren die vandaag aan deze patiënten kunnen worden aangeboden. Wij zijn van mening dat het van het grootste belang is dat niet alleen de huisarts over een actuele kennis van dit onderwerp beschikt, maar dat ook met de longarts wordt samengewerkt om een goede therapietrouw van de patiënt en een vroegtijdige opsporing en behandeling van eventuele complicaties te bereiken.
Van de interstitiële ziekten is idiopathische longfibrose (IPF) de meest relevante vanwege de frequentie en ernst ervan. Anderzijds is de behandeling ervan het model waarop de behandeling van bijna al deze ziekten is gebaseerd, met uitzondering van die waarin een bekend etiologisch agens kan worden gevonden, aangezien in deze gevallen de eliminatie van dit agens de fundamentele basis van de therapie zal zijn. Wij zullen ons daarom concentreren op een herziening van de huidige richtsnoeren voor de behandeling van IPF. We zullen eerst kort de klinische kenmerken bespreken en vervolgens de huidige behandeling van IPF in detail ontwikkelen.
CLINISCHE BESCHRIJVING
IPF vertegenwoordigt waarschijnlijk het meest agressieve prototype van de diffuse interstitiële pneumopathieën. De prevalentie is door sommige auteurs geschat op 5 gevallen per 100.000 inwoners1 , hoewel deze in werkelijkheid onbekend is. De pathogenese houdt verband met veranderingen in de complexe routes van het immuunsysteem2,3. 2,3 Daarom worden meestal immunosuppressieve middelen gebruikt.
De meest voorkomende klinische presentatie is een dyspneu die sluipend begint en gepaard gaat met droge hoest, wat voor de patiënt het meest storende symptoom kan zijn vanwege het paroxysmale en ademloze karakter. In het algemeen is dyspneu aanvankelijk onopvallend en een hoge verdenkingsindex is noodzakelijk voor een vroege diagnose. Soms is het beloop snel na het begin en leidt het binnen enkele maanden tot onomkeerbare ademhalingsinsufficiëntie, zoals kenmerkend was voor de eerste beschrijvingen door Hamman en Rich. Meestal is de aandoening echter al maanden aanwezig tegen de tijd dat de patiënt zijn of haar eerste bezoek aflegt.
Bij lichamelijk onderzoek is fijn, droog gekraak (velcro rales) zeer kenmerkend en kan in meer dan 80% van de gevallen worden gehoord. In gevorderde gevallen kan dyspneu bij lichte inspanning of zelfs lipcyanose worden gezien. Acropaquias kunnen worden gezien in ongeveer 30-50% van de gevallen. Het vermoeden van IPF wordt versterkt wanneer op röntgenfoto’s van de borstkas en CT-scan tekenen verschijnen zoals een reticulair patroon, gebieden met grondglasinfiltratie en verminderd longvolume. Ten slotte zal het functioneel onderzoek het bestaan van een restrictief patroon bevestigen en zal arteriële bloedgasanalyse het bestaan van hypoxemie bevestigen. In twijfelgevallen is het aantonen van hypoxemie bij inspanning van grote waarde.
De definitieve bevestiging van de diagnose kan alleen worden gedaan door een longbiopsie. De realiteit is echter dat zelfs in veel van de gepubliceerde reeksen de gevallen zonder histologische diagnose overheersen. Onlangs heeft de American Thoracic Society (ATS) voorgesteld dat het bestaan van al deze criteria betekent dat, bij afwezigheid van blootstelling aan geneesmiddelen, milieu-elementen of bindweefselziekten, de diagnose IPF hoogstwaarschijnlijk kan worden gesteld zonder longbiopsie4 in gevallen waarin een longbiopsie een onaanvaardbaar risico zou vormen.
Usuele interstitiële pneumonie (UIP) is het histopathologische patroon dat deze patiënten identificeert4. Die met patronen van desquamatieve interstitiële pneumonie, interstitiële ziekte geassocieerd met respiratoire bronchiolitis, aspecifieke interstitiële pneumonie, lymfoïde interstitiële pneumonie, acute interstitiële pneumonie, en bronchiolitis obliterans met organiserende pneumonie worden beschouwd als verschillende entiteiten.
Als de diagnose en de klinische ernst eenmaal zijn vastgesteld, hebben de meeste patiënten behandeling nodig, omdat het een dodelijke ziekte is zonder spontane remissie. De reactie op de behandeling is slecht. Vijf-jaars overleving is 50%5,2,6-8. De behandeling duurt ongeveer 3-6 maanden voordat de effectiviteit is vastgesteld. Verslagen over de behandeling van deze ziekten zijn schaars: ongecontroleerde therapeutische proeven, meestal met kleine aantallen patiënten9.
Ondanks het onvermijdelijke verraderlijke en onomkeerbare verloop van de ziekte is therapeutisch nihilisme niet gerechtvaardigd. Er zijn voorbeelden van onbehandelde fibrose met langzame progressie die leidt tot onomkeerbare fibrose die niet reageert op behandeling of die een snel progressieve fase ontwikkelt. Ze moeten waarschijnlijk allemaal behandeld worden op het moment van de presentatie. Patiënten met contra-indicaties voor behandeling of patiënten zonder aanwijzingen voor functionele stoornissen kunnen echter met tussenpozen van 3-6 maanden een follow-upprogramma volgen. De behandeling van oudere patiënten met een overwegend fibroserijke histologie en ernstige longfunctiestoornissen is eveneens moeilijk. Deze patiënten krijgen een korte behandeling van 3-6 maanden om de omkeerbaarheid vast te stellen. Slechts weinigen van hen zullen een positieve reactie vertonen.
Het is vaak moeilijk vast te stellen of de behandeling het gewenste effect sorteert. Subjectieve verbetering treedt vaak op (bij tot 70% van de patiënten). Objectieve verbetering treedt op bij 20-30%. De meest gebruikte parameters om functionele (objectieve) verbetering te definiëren zijn een toename van de totale longcapaciteit (TLC) of de vitale capaciteit (VC), een toename van de diffusiecapaciteit en een vermindering of normalisering van de SaO2-daling tijdens inspanning4,10-12.
Behandeling
Corticosteroïden
Corticosteroïden vormen de belangrijkste pijler van de behandeling van IPF, hoewel het succes ervan vrij gering is: een gunstige respons komt voor bij een derde van de patiënten. De reden voor het gebruik ervan is de onderdrukking van chronische alveolitis. Men denkt dat ze werken door de migratie van neutrofielen en lymfocyten in de long te onderdrukken, door de concentratie van immunocomplexen te verlagen, de alveolaire macrofaagfunctie te veranderen en de hechting van neutrofielen aan endotheliale oppervlakken te beïnvloeden.
Het is niet duidelijk waarom sommige patiënten wel en andere niet reageren. Het kan verband houden met de aanwezigheid van oppervlakte-receptoren op longparenchymcellen. Een kort tijdsverloop van de symptomen, milde functionele en radiologische stoornissen, het vrouwelijk geslacht en een jongere leeftijd van de patiënten lijken samen te hangen met een beter resultaat.
Hoewel het de meest gebruikte behandeling is, is de werking op lange termijn niet bevestigd door een prospectieve, een-blinde, placebo-gecontroleerde studie.
Izumi et al analyseerden de overleving van 222 patiënten en ontdekten dat er na 10 jaar geen verschil in overleving was tussen degenen die steroïden hadden gekregen en onbehandelde patiënten13. Wanneer een patiënt met IPF zich echter presenteert met een verminderde longfunctie of symptomen die het normale leven belemmeren, is het redelijk om een poging tot behandeling met corticosteroïden te ondernemen. In feite is spontane verbetering van de ziekte zeer zeldzaam, en als besloten wordt de start van de behandeling uit te stellen, moet dit ten koste gaan van een nauwgezette controle van de patiënt, zodat de behandeling kan worden gestart zodra verslechtering duidelijk wordt.
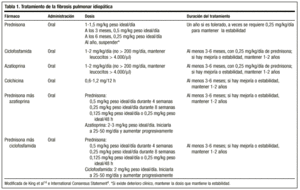
De klassieke behandeling begint met prednison 1,5-2 mg/kg ideaal gewicht/dag als eenmalige dosis in de ochtend, waarbij een dosis van 100 mg niet mag worden overschreden. Na 2-3 maanden wordt de patiënt opnieuw geëvalueerd met radiografie en functieonderzoek. Als de patiënt reageert, wordt prednison afgebouwd (1-2 mg/week) tot een onderhoudsdosis van 0,5 mg/kg ideaal gewicht/dag, die daarna kan worden verlaagd. Als er geen reactie is, wordt een immunosuppressief medicijn toegevoegd. In het algemeen is het onwaarschijnlijk dat de behandeling kan worden stopgezet, en doses van 0,25 mg/kg ideaal gewicht/dag worden gewoonlijk gehandhaafd.
Onlangs hebben de Amerikaanse en Europese Thoracale Genootschappen, om de bijwerkingen van dit geneesmiddel te vermijden en betere overlevingskansen te verkrijgen, een consensus bereikt om de behandeling te beginnen met een combinatie van prednison of een ander corticosteroïd in gelijkwaardige doses en een immunosuppressivum (azathioprine of cyclofosfamide)4: prednison 0,5 mg/kg ideaal gewicht/dag gedurende 4 weken; 0,25 mg/kg ideaal gewicht/dag gedurende 8 weken, en dan geleidelijk afbouwen tot 0,125 mg/kg ideaal gewicht/dag of 0,25 mg/kg ideaal gewicht/48 uur.
Er zijn geen richtlijnen voor de duur van de behandeling; de minimumduur is één jaar. Het is mogelijk dat de ziekte na maanden of jaren opnieuw de kop opsteekt, in welk geval een behandeling met dezelfde middelen moet worden ingesteld. In ieder geval moet de respons op de behandeling altijd worden beoordeeld in termen van objectieve verbetering en moet de mogelijkheid om de patiënt chronisch op een dosis te houden die stabiliteit garandeert, worden gereserveerd voor die gevallen waarin verbetering optreedt met corticosteroïden, verslechtering optreedt bij afbouwen en opnieuw verbetering optreedt bij opnieuw verhogen van de dosis. De voor elke patiënt vereiste dosis kan alleen van geval tot geval worden bepaald, afhankelijk van de reactie op veranderingen in de dosis. Veel auteurs achten levenslange behandeling noodzakelijk.14
Wanneer de ziekte aanvankelijk ernstig is, zijn hoge doses corticosteroïden voorgesteld. Keogh et al hebben bij patiënten met ernstige ziekte intermitterend hoge doses parenterale corticosteroïden gebruikt: intraveneus methylprednisolon, 2 g eenmaal per week, plus oraal prednison, 0,25 mg/kg/dag. De reden hiervoor is dat zij een meer uitgesproken effect hebben op neutrofiele alveolitis (geassocieerd met een slechte prognose) dan lage-dosis steroïden15. Dit werk lijdt echter aan enkele tekortkomingen, zoals de korte follow-up van 6 maanden, het kleine aantal patiënten en de lagere dan standaarddoses prednison9. Een initiële behandeling met hoge doses intraveneuze corticosteroïden (methylprednisolon 250 mg/6 u) is gebruikt in een poging om de ziekte zo vroeg mogelijk onder controle te krijgen wanneer deze snel progressief is. Er zijn geen gecontroleerde studies om vast te stellen of deze methoden doeltreffend zijn; hogere doses lijken echter een grotere doeltreffendheid te hebben, hoewel de bijwerkingen ook groter zijn.14.
CITOTOXISCHE AGENTIA
Zijn klassiek gebruikt als alternatief voor corticosteroïden wanneer deze niet doeltreffend waren, en ook wanneer de bijwerkingen verdere toediening ervan onmogelijk maken. In de bij uitstek praktische geest van dit artikel zullen wij de middelen bespreken die in de klinische praktijk hun nut hebben bewezen, hoewel wij, zoals in het geval van de corticosteroïden, niet beschikken over lange prospectieve en gerandomiseerde reeksen die ons in staat stellen precies te weten wat hun rol bij de behandeling van IPF is.
Azathioprine
Het werkingsmechanisme is gebaseerd op de substitutie van purines in de desoxyribonucleïnezuursynthese, waardoor adenine deaminase wordt geremd en lymfocyten worden geïnactiveerd. Andere effecten zijn onderdrukking van de activiteit van natuurlijke killercellen, de productie van antilichamen en antilichaam-afhankelijke cellulaire cytotoxiciteit.
De aanbevolen dosis is 2 mg/kg/dag, niet meer dan 200 mg/dag, gedurende een periode van 3-6 maanden. De toediening wordt gestart met een dosis van 50 mg/dag en, indien goed verdragen, wordt de dosis met tussenpozen van 1 tot 2 weken verhoogd tot de geplande dosis is bereikt.
De belangrijkste bijwerkingen zijn hematologisch: leukopenie, anemie, trombocytopenie, rode cel aplasie en pancytopenie. Het is noodzakelijk het aantal leukocyten te controleren en indien dit onder 4.000/μl daalt, moet de azathioprine worden gestaakt totdat dit aantal weer stijgt. Bovendien kunnen gastro-intestinale ongemakken zoals misselijkheid en braken, maagzweer, diarree en lichte verhoging van leverenzymen optreden. Over het geheel genomen vereisen bijwerkingen in 20-30% van de gevallen het staken van azathioprine.
Raghu et al vergeleken de werkzaamheid van hooggedoseerde glucocorticoïden versus azathioprine in combinatie met laaggedoseerde glucocorticoïden. Het gebruik van dit middel ging gepaard met een bescheiden verbetering van de ademhalingsfunctieparameters en een grotere overleving17.
Cyclophosphamide
Is een alkylerend middel. Het werkingsmechanisme is de uitputting van lymfocyten en onderdrukt dus de lymfocytenfunctie. De aanbevolen dosis is 2 mg/kg/dag eenmalig, oraal, meestal in combinatie met 0,25 mg/kg/dag oraal prednison, niet meer dan 200 mg/dag gedurende ten minste 3 maanden en maximaal 9-12 maanden.
De belangrijkste bijwerking is leukopenie. Het totale aantal leukocyten moet meer dan 3000 per ul bedragen of het aantal neutrofielen moet meer dan 1500 per ul bedragen. Andere complicaties zijn trombocytopenie, hematurie secundair aan hemorragische cystitis, anorexie, misselijkheid en braken, beenmergsuppressie, azoöspermie en amenorroe, infectie en hematologische maligniteit.
Kolb et al onderzochten 18 patiënten met IPF om de werkzaamheid en veiligheid van gepulseerde cyclofosfamide therapie te bepalen. Cyclofosfamide werd intermitterend gegeven (1-1,3 g/maand) samen met oraal prednisolon gedurende een jaar. Na deze periode werd de cyclofosfamide gestaakt en werden de bereikte doses corticosteroïden voortgezet8 . Zij werden gedurende ten minste 14 maanden gevolgd, waarbij de QoL, TLC, FEV1, CO-overdrachtscapaciteit (DLCO), SaO2, alsmede de symptomen werden gecontroleerd. Bij één patiënt moest de behandeling worden gestaakt wegens terugkerende pneumonieën; 11 patiënten werden als responders beschouwd (5 verbeterd en 6 gestabiliseerd) en 6 verslechterden. De toxiciteit op lange termijn van cyclofosfamide is afhankelijk van de cumulatieve doses. Intermitterende doses zijn even doeltreffend en worden beter verdragen dan dagelijkse doses. Responders hadden een hoger percentage lymfocyten in BAL. Een andere opmerkelijke bevinding is dat de aanvankelijke QOL van responders significant beter was als gevolg van een kortere tijd tot progressie, wat suggereert dat deze behandeling vroeg moet worden gestart.
Baughman en Lower behandelden 33 IPF-patiënten met 1.000-1.800 mg cyclofosfamide/2 weken gedurende 18 maanden. Degenen die 18 maanden overleefden, hadden een significante verbetering in VCF die het volgende jaar gehandhaafd bleef. Vermindering van prednisolon was mogelijk van 32 tot 4 mg/dag16.
Penicillamine
Antifibrosant middel. Er is gesuggereerd dat het de ontwikkeling van longfibrose remt door de vorming van intra- en intermoleculaire covalente bindingen van collageenvezels te belemmeren. Er zijn enkele gevallen van bindweefselziekten met geassocieerde longfibrose beschreven waarin het enige werkzaamheid heeft gehad, maar er zijn geen gecontroleerde studies in grote series patiënten, zodat het momenteel niet als een geldige alternatieve behandeling kan worden beschouwd. Daarom is verder onderzoek nodig voordat een aanbeveling kan worden gedaan.
Colchicine
Vermindert de collageenafscheiding en verhoogt de collageenolytische activiteit. Het heeft ook een ontstekingsremmend effect. Studies zijn nodig om de rol ervan bij de behandeling van deze ziekte nauwkeuriger te bepalen, maar er zijn studies waarin de werkzaamheid ervan wordt toegeschreven aan het feit dat zij vergelijkbaar is met die van corticoïden, en de bijwerkingen zijn niet ernstig. Het wordt aanbevolen in een dosis van 0,6 mg, eenmaal of tweemaal per dag, bij patiënten die refractair zijn voor corticosteroïden, alleen of in combinatie met immunosuppressieve middelen. Hoewel er auteurs zijn die het in de eerste lijn van behandeling plaatsen, ontbreekt het ons momenteel aan gegevens om het als zodanig vast te stellen (tabel 1).
PULMONAIRE TRANSPLANT
De gevallen waarin de ziekte voortschrijdt zonder dat op de behandeling wordt gereageerd en uiteindelijk tot ademhalingsfalen leidt, zijn helaas het talrijkst bij deze ziekte. Er is momenteel geen uitzicht op medische behandeling voor deze patiënten, behalve palliatie. Tegen deze sombere achtergrond is in de jaren tachtig de mogelijkheid van longtransplantatie ontstaan, die zich zodanig heeft ontwikkeld dat het nu een therapeutische optie is die moet worden beschouwd als een instrument voor klinisch gebruik en niet langer beperkt is tot het experimentele gebied. De meest geschikte indicatie is bij mensen jonger dan 65 jaar, bij afwezigheid van progressieve en onomkeerbare ziekte in andere organen, en van onopgeloste actieve infectie of infectie met multiresistente kiemen. Adequate persoonlijke aanleg en voedingstoestand zijn eveneens vereist. Patiënten met een CPT van minder dan 60% van de verwachte waarde of een longdiffusiecapaciteit van minder dan 40% hebben een overlevingsduur van minder dan 2 jaar. In deze mate van disfunctie hebben zij zuurstoftherapie nodig en desatureren zij gemakkelijk bij minimale inspanning. Longtransplantatie moet in deze gevallen worden overwogen, zodra zuurstoftherapie noodzakelijk wordt en een onstuitbare progressie van de ziekte wordt vastgesteld. Het type transplantatie dat de voorkeur geniet is de transplantatie van één long, waarbij de transplantatie van twee longen wordt gereserveerd voor gevallen waarin twijfel bestaat over het gedrag van de overblijvende long. Als de patiënt jonger is dan 50 jaar en rechterhartfalen heeft, kan cardiopulmonale transplantatie worden overwogen. In deze situatie is de ziekte echter zeer ver gevorderd en is het onwaarschijnlijk dat de wachttijd kan worden doorstaan18,19. Naar verwachting zal de overleving na de transplantatie het eerste jaar 80% bedragen en 50-60% na 5 jaar, wat de prognose voor een patiënt met IPF die respiratoir falen heeft bereikt, aanzienlijk verbetert.
EVALUATIE VAN DE RESPONS OP DE BEHANDELING
Bij afwezigheid van complicaties kan het tot 6 maanden duren voordat er enige respons op de behandeling optreedt. Na deze tijd, als de patiënt is verbeterd of stabiel is, moeten dezelfde doses worden gehandhaafd. Als deze verergerd zijn, moet worden overgeschakeld op een alternatieve therapie (een ander immunosuppressief middel of zelfs transplantatie). Na 12 maanden moet het op dezelfde wijze opnieuw worden geëvalueerd en na 18 maanden geïndividualiseerd naar gelang van de respons. Se recomienda continuar indefinidamente sólo en aquellos pacientes con mejoría continua o estabilización (tabla 2).

Una respuesta favorable se define por dos o más de los siguientes criterios en dos visitas consecutivas:
1. Mejoría de los síntomas, sobre todo en el grado de esfuerzo requerido para que el paciente deba parar cuando hace un esfuerzo o un descenso en la frecuencia o gravedad de la tos.
2. Reducción de las alteraciones radiológicas.
3. Mejoría funcional definida por dos o más de los siguientes parámetros:
Aumento >= 10% en la TLC o CV (o >= 200 ml).
Aumento >= 15% en DLCO o >= 3 ml/min/mmHg.
Mejoría o normalización de la SaO2 o pO2 (aumento >= 4% o >= 4 mmHg) durante un prueba de esfuerzo cardiorrespiratoria.
Una respuesta estable (también considerada favorable) se define por dos o más de los siguientes criterios en dos visitas consecutivas:
1. Cambio de
2. Cambio de
3. Sin cambios en la SaO2 o pO2 (aumento
Un fracaso del tratamiento se define por dos o más de los siguientes criterios en dos visitas consecutivas:
1. Aumento de los síntomas, sobre todo disnea o tos.
2. Aumento de las alteraciones radiológicas, especialmente imágenes en panal o signos de hipertensión pulmonar.
3. Deterioro funcional definido por dos o más de los siguientes parámetros:
Descenso >= 10% en la TLC o CV (o >= 200 ml).
Descenso >= 15% en DLCO o >= 3 ml/min/mmHg.
Verergering van de SaO2 (= 4%) of verhoging van de alveolair-arteriële gradiënt (AapO2) (= 4 mmHg) in rust of tijdens een cardiorespiratoire inspanningstest.
CONCLUSIES
Idiopathische longfibrose is een fatale ziekte zonder spontane remissie en slecht reagerend op behandeling. Studies over dit laatste zijn schaars en betreffen een klein aantal patiënten. Corticosteroïden die gedurende ten minste één jaar in verschillende doses worden gehandhaafd, vormen de meest aanvaarde behandeling, alleen of in combinatie met andere immunosuppressieve middelen. Onlangs is combinatietherapie bepleit door de American Thoracic en European Respiratory Societies4 . Anti-fibrose middelen worden onderzocht met de bedoeling te voorkomen dat het fibrotisch proces zich vastzet en dus onomkeerbaar wordt, aangezien veel patiënten in een laat stadium worden gediagnosticeerd. Gezien de overlevingspercentages en de evolutie moet longtransplantatie worden overwogen zodra patiënten niet meer kunnen worden behandeld en zuurstoftherapie nodig hebben. Dit alles wijst op de noodzaak van multicenterstudies met het oog op de rekrutering van een voldoende aantal patiënten om de doeltreffendheid van de toegediende behandelingen te beoordelen, alsook om de natuurlijke geschiedenis van de ziekte en de reële incidentie en prevalentie vast te stellen4,20. Anderzijds wordt aanbevolen de ziekte in een vroeg stadium op te sporen en met de behandeling ervan te beginnen om een hoger overlevingspercentage te bereiken.