Veel van de methoden voor het visualiseren en interpreteren van genexpressie data kunnen worden gebruikt voor zowel microarray als RNA-seq experimenten. Enkele van de meest gebruikte methoden worden hieronder besproken.
Heatmaps en clustering
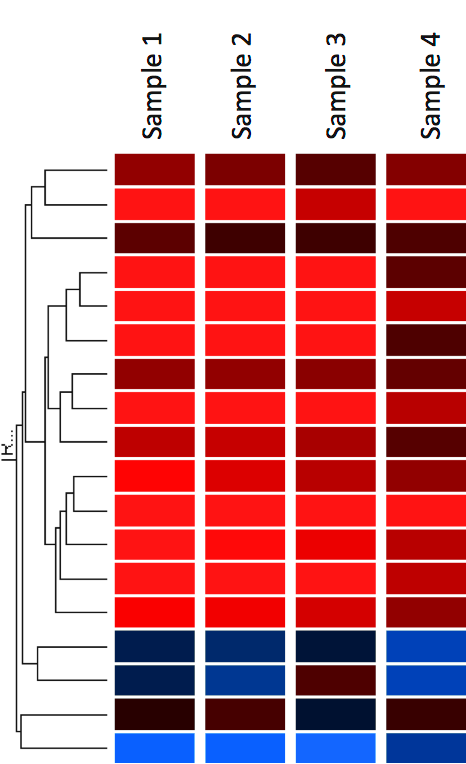
Een veelgebruikte methode voor het visualiseren van genexpressiegegevens is deze weer te geven als een heatmap (figuur 12). De heatmap kan ook gecombineerd worden met clustermethoden die genen en/of stalen groeperen op basis van de gelijkenis van hun genexpressiepatroon. Dit kan nuttig zijn voor het identificeren van genen die algemeen gereguleerd zijn, of biologische handtekeningen die geassocieerd zijn met een bepaalde aandoening (bv. een ziekte of een milieuconditie) (4).
In heat maps worden de gegevens weergegeven in een raster waarbij elke rij een gen voorstelt en elke kolom een monster. De kleur en intensiteit van de vakken wordt gebruikt om veranderingen (geen absolute waarden) van genexpressie weer te geven. In het voorbeeld hieronder vertegenwoordigt rood opwaarts gereguleerde genen en blauw neerwaarts gereguleerde genen. Zwart staat voor onveranderde expressie.
Genensetverrijkingsanalyse en padanalyse
Een veelgebruikte aanpak voor de interpretatie van genexpressiegegevens is genensetverrijkingsanalyse op basis van de functionele annotatie van de differentieel tot expressie gebrachte genen (figuur 13). Dit is nuttig om uit te zoeken of de differentieel tot expressie komende genen geassocieerd zijn met een bepaald biologisch proces of een bepaalde moleculaire functie.
De Gene Ontology, die gestandaardiseerde annotatie van genproducten bevat, wordt vaak voor dit doel gebruikt. Het werkt door de frequentie van individuele annotaties in de genenlijst (b.v. differentieel tot expressie komende genen) te vergelijken met een referentielijst (meestal alle genen op de microarray of in het genoom). Verrijking van biologische paden geleverd door KEGG, Ingenuity, Reactome of WikiPathways kan worden uitgevoerd op een vergelijkbare manier (12,13).
Populaire hulpmiddelen voor gen set verrijking en pathway analyse omvatten:
- DAVID (gratis online tool)
- GSEA (gratis)
- Ingenuity (licentie vereist)
- Reactome (gratis)

Netwerkanalyse
Netwerkanalyse is een aanvulling op de analyse van paden en kan worden gebruikt om te laten zien hoe belangrijke componenten van verschillende paden op elkaar inwerken. Dit kan nuttig zijn voor het identificeren van regulerende gebeurtenissen die meerdere biologische processen en paden beïnvloeden (12,13).