- Introduction
- Materialen en Methoden
- Bemonstervoorbereiding
- MinION-bibliotheekbereiding en sequentiebepaling
- Genomics Analysis
- Results
- MinION Sequencing Data en assemblage van virale genomen
- Bereik en bron van kruisbesmetting
- Discussie
- Author Contributions
- Funding
- Conflict of Interest Statement
- Acknowledgments
- Aanvullend materiaal
- Voetnoten
Introduction
Metagenomic sequencing heeft de potentie om onpartijdige identificatie van pathogenen uit een klinisch monster mogelijk te maken. Het houdt de belofte in om te dienen als een enkele en universele assay voor de diagnostiek van infectieziekten direct uit monsters zonder de noodzaak van a priori kennis (Bibby, 2013; Miller et al., 2013; Schlaberg et al., 2017). Naast identificatie van pathogeensoorten zouden brede en diepe metagenomische sequentiegegevens informatie kunnen opleveren die relevant is voor het bepalen van behandeling en prognose, het opsporen van uitbraken en het volgen van infectie-epidemiologie (Greninger et al., 2010; Yang et al., 2011; Qin et al., 2012; Loman et al., 2013). Next-generation sequencing (NGS) platforms kunnen een enorme doorvoer van gegevens produceren tegen een bescheiden kostprijs, maar de toepassing ervan in de klinische diagnostiek en de volksgezondheid is beperkt door complexiteit, traagheid en kapitaalinvestering.
De MinION is een palm-formaat, real-time, single-molecule genoom sequencer ontwikkeld door Oxford Nanopore Technologies (ONT). Het compacte formaat en het realtime karakter van de MinION zouden de toepassing van metagenomische sequencing in point-of-care testen voor infectieziekten kunnen vergemakkelijken, zoals blijkt uit verschillende proof-of-conceptstudies, waaronder de identificatie van Chikungunya (CHIKV), Ebola (EBOV) en hepatitis C-virus (HCV) uit menselijke klinische bloedmonsters zonder doelverrijking (Greninger et al, 2015), en detectie van bacteriële pathogenen uit urinemonsters (Schmidt et al., 2016) en ademhalingsmonsters, zonder de noodzaak van voorafgaande kweek (Pendleton et al., 2017).
De gegevensdoorvoer van MinION is sterk toegenomen sinds de release in 2015, waarbij elke verbruikbare flowcel nu tot 10-20 Gb aan DNA-sequentiegegevens genereert. Dit stelt gebruikers in staat om efficiënter gebruik te maken van de flowcel (en de kosten te verlagen) door multiplexing van meerdere monsters in een enkele sequencing run. ONT heeft PCR-vrije barcodesets ontwikkeld waarmee tot 12 monsters kunnen worden gemultiplexed.
Het opsporen van influenza A-virus in meerdere respiratoire monsters zou een diagnostische toepassing van een multiplexed MinION-sequencingassay kunnen zijn. Bij directe sequencing van monsters met een potentieel breed spectrum aan virale titers is het echter belangrijk om rekening te houden met mogelijke kruiscontaminatie, zowel tijdens de voorbereiding van de bibliotheek als tijdens de bioinformatische demultiplexing van de barcode na de sequencing. Hier presenteren we een unieke MinION sequencing dataset en de resultaten van het onderzoek naar de omvang en de bron van cross-barcode contaminatie in multiplex sequencing.
Materialen en Methoden
We gebruikten een fret nasale wassen monster geïnfecteerd met influenza A-virus als een voorbeeld en ook spiked twee aliquots van negatieve nasale wassen monsters van niet-geïnfecteerde fret (pre-bestaande ongebruikte voorraden van een niet-gerelateerde studie) met dengue en chikungunya virussen afzonderlijk. Geen van beide virussen zijn relevant voor klinische diagnostiek in respiratoire monsters, maar fungeren hier als duidelijke, afzonderlijke markers voor de beoordeling van kruisbesmetting van monsters. De sequencingbibliotheken voor elk monster werden parallel bereid, samen met een negatieve neusspoelcontrole, voorzien van een streepjescode, en individueel gesequenced. Vervolgens hebben we een aliquot van de sequencingbibliotheken gepoold en multiplex MinION-sequencing uitgevoerd. Lezingen van de vier afzonderlijke runs (aangeduid als “CHIKV”, “DENV”, “FLU-A” en “Negatief”) en de multiplex run (aangeduid als “Multiplexed”) werden vervolgens geanalyseerd om de omvang en bron van kruisbesmetting te onderzoeken.
Bemonstervoorbereiding
De projectvergunning werd beoordeeld door de lokale AWERB (Animal Welfare and Ethics Review Board) en werd vervolgens verleend door het Home Office. RNA werd geëxtraheerd, met behulp van de QIAamp virale RNA kit (Qiagen) volgens de instructies van de fabrikant, uit fretten neusspoeling met influenza A (H1N1) virus (A/California/04/2009) en een pool van negatieve neusspoelingsmonsters. Aliquots van het negatieve monsterextract werden gespiked met dengue (DENV) (stam TC861HA, GenBank: MF576311) of CHIKV (stam S27, GenBank: MF580946.1) viraal RNA uit de National collection of Pathogenic Viruses1. Monsters werden DNase behandeld met TURBO DNase (Thermo Fisher Scientific, Waltham, MA, Verenigde Staten) en gezuiverd met behulp van de RNA Clean & ConcentratorTM-5 kit (Zymo Research). cDNA werd bereid en geamplificeerd met behulp van een Sequence-Independent-Single-Primer-Amplification methoden (Greninger et al., 2015) gewijzigd zoals eerder beschreven (Atkinson et al., 2016). Geamplificeerd cDNA werd gekwantificeerd met behulp van de Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA, Verenigde Staten), en 1 ug werd gebruikt als input voor elke MinION bibliotheek voorbereiding, met uitzondering van de negatieve controle waar het gehele monster (32 ng) werd gebruikt.
MinION-bibliotheekbereiding en sequentiebepaling
Ligatie Sequencing Kit 1D (SQK-LSK108) en Native Barcoding Kit 1D (EXP-NBD103) werden gebruikt volgens de ONT-standaardprotocollen, met de uitzondering dat slechts een barcode werd opgenomen in elk van de vier bibliotheekpreparaten. Elke bibliotheek werd uitgevoerd op een individuele flowcel en een vijfde gepoolde bibliotheek werd gemaakt door het combineren van de vier individueel gebarcodeerde bibliotheken. De bibliotheken werden gesequenced op R9.4 flowcellen. De studieopzet wordt getoond in figuur 1.
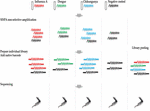
FIGUUR 1. Overzicht van de onderzoeksopzet. RNA werd geëxtraheerd uit vier monsters, waaronder een frettenneuswasmonster dat met influenza A-virus was geïnfecteerd, twee negatieve frettenneuswasmonsters die met dengue- en chikungunya-virussen waren besmet, en een negatieve frettenneuswascontrole. cDNA werd bereid en geamplificeerd met behulp van een sequentie-onafhankelijke singel-primer-amplificatiemethode. De sequencingbibliotheken voor elk monster werden parallel bereid, van een streepjescode voorzien en op afzonderlijke flowcellen gesequeneerd. Multiplex-sequencing werd ook uitgevoerd door de vier individuele bibliotheken samen te voegen. De resultaten van de vier afzonderlijke sequenceruns en de multiplexsequenceruns werden geanalyseerd om de omvang en bron van kruisbesmetting met streepjescodes bij multiplexsequencing te bepalen.
Genomics Analysis
Resultaten werden met Albacore v2.1.7 (ONT) met streepjescode-demultiplexing als basecall gebruikt. Lezingen van elke sequencing run werden in kaart gebracht op genomische sequenties van elk virus met behulp van Minimap2 (Li, 2018). Het aantal gelezen in kaart gebracht om referentie werd geteld met behulp van Pysam2. De novo assemblage werd uitgevoerd met Canu v1.7 (Koren et al., 2017), en het resulterende conceptgenoom werd opgepoetst met Nanopolish (Mongan et al., 2015) met de signaalniveaugegevens.
Om strikte barcode demultiplexing van de multiplex MinION-sequencinggegevens mogelijk te maken, voerden we twee analyserondes uit met Porechop (v0.2.23). Aanwezigheid van adaptersequentie in het midden van een lees is een handtekening van chimera. We gebruikten Porechop om elke lezing te onderzoeken en die hebben midden regio delen >75% identiteit met adapter sequentie werden geïdentificeerd als chimera gelezen. In Porechop, zetten we de “-middle_threshold” optie en kiezen een drempel van 75. In de tweede ronde, gebruikten we Porechop om de barcode sequentie op te zoeken aan zowel het begin als het einde van een lees; leeswaarden werden alleen toegewezen als dezelfde barcode werd gevonden aan twee uiteinden. We hebben de “-require_two_barcodes” optie in Porechop ingesteld en de drempel voor de barcode score op 70 gesteld. Om potentiële handtekening van chimerische leest vinden, we onderzochten de gelezen huidige signalen opgeslagen in de FAST5 bestand door MinION sequencer. Huidige signalen werden geëxtraheerd met behulp van ONT fast5 API4 en uitgezet met behulp van ggplot2 geïmplementeerd in R5 voor een vergelijking van chimerische en niet-chimerische reads.
Results
De doorvoer van elke MinION sequencing run varieerde als gevolg van verschillen in looptijd. Een maximum aantal ∼ 2,4 M leest werd bereikt door de multiplexed sequencing run en de individuele CHIKV run, als gevolg van langere looptijden (Supplementary Table S1). Leest van de spiked virus goed voor 96% van de gegevens in de individuele CHIKV en DENV sequencing runs, en 78% voor de FLU-A monster (tabel 1). Het percentage virale gegevens binnen elk monster met streepjescode in de sequencinggegevens met multiplex ligt dicht bij dat in de gegevens van de individueel uitgevoerde monsters (tabel 2). Elk viraal genoom had een ultrahoge (>8.000) gemiddelde dekkingsdiepte in de individuele en multiplex sequencing gegevens, en de novo assemblage was in staat om bijna volledige genomen voor alle drie virussen met 99.De de novo assemblage was in staat bijna volledige genomen voor alle drie virussen te verkrijgen met 99,9% identiteiten vergeleken met de GenBank-referentie.

TABLE 1. Samenvatting van de resultaten van mapping en de novo assemblage voor gegevens van MinION-sequencing van afzonderlijke bibliotheken.

TABLE 2.
Bereik en bron van kruisbesmetting
Elk monster was voorzien van een streepjescode en zowel individueel als multiplex gesequenced, zodat we de prestaties van de demultiplexing van de streepjescode van Albacore konden onderzoeken. In de individueel gesequenteerde monstergegevens zouden we verwachten dat er slechts één native barcode aanwezig is. Voor CHIKV (barcode NB01), DENV (NB09), en FLU-A (NB10) individuele sequencing runs, vonden we dat 86, 109, en 17 leest, respectievelijk, werden toegewezen aan barcode bins niet verwacht aanwezig te zijn in de bibliotheek (vertegenwoordigen 0.0036, 0.0129, en 0.001% van het totaal leest). In de multiplex sequencing gegevens, 41 leest (0.0016%) werden toegewezen aan barcodes niet opgenomen in de experimenten (dat wil zeggen, een barcode anders dan NB01, NB05, NB09, of NB10). We hebben deze gedefinieerd als verkeerd toegewezen leest (figuur 2A).
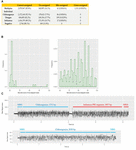
FIGUUR 2. (A) Overzicht van het aantal en percentage correct toegewezen, niet toegewezen, verkeerd toegewezen en kruiselings toegewezen gelezen in elke sequencingreeks. Niet-toegewezen verwijst naar leest die niet kan worden toegewezen aan een bins door Albacore als gevolg van een barcode score lager dan 60, verkeerd toegewezen verwijst naar leest die werden toegewezen aan barcode bins niet opgenomen in dit experiment, en cross-toegewezen verwijst naar leest die werden toegewezen aan de onjuiste barcode bins; (B) verdeling van barcode scores gemeld door Albacore voor verkeerd toegewezen leest en cross-toegewezen leest in de multiplex sequencing gegevens; (C) vergelijking van ruwe signaal van een chimere en een correct toegewezen lezen. Het signaal van chimerische lezingen vertoont een stagnatiesignaal en een enorm pieksignaal in het midden van de lezing.
Om mogelijke laboratoriumcontaminatie in de sequencingbibliotheekvoorbereiding te onderzoeken, hebben we alle lezingen van elke afzonderlijke run gemapt tegen de genoomsequenties van alle drie virussen. Geen enkele lees was afkomstig van een genoom dat in een andere bibliotheek was geprepareerd, wat erop wijst dat er geen in-vitro-verontreiniging heeft plaatsgevonden. De multiplex sequencing-bibliotheek werd bereid door pooling van de afzonderlijke, niet-verontreinigde bibliotheken na de ligatie van zowel barcode als adapter. Echter, mapping resultaten tonen 1,311 (0.0543%) leest gekoppeld aan de onjuiste doelgenoom, wat impliceert dat ze kruislings toegewezen aan de verkeerde barcode bins (later aangeduid als “cross-toegewezen leest”), ondanks het feit dat de multiplexed sequencing bibliotheek werd gepoold met individuele bibliotheken toonde geen kruislings toegewezen leest op alle. We veronderstelden dat verkeerd toegewezen en cross-toegewezen leest waren te wijten aan een lage barcode score, en onderzocht de barcode scores van deze leest. De meeste van de verkeerd toegewezen leest had een barcode score <70, echter, cross-toegewezen leest had meer uiteenlopende scores variërend van 60 tot bijna 100 (Figuur 2B). Dit resultaat suggereert dat verkeerd toegewezen en kruis-toegewezen leest afkomstig zijn van verschillende bronnen. We blasten de kruis-toegewezen leest aan een kleine database die de genoomsequenties van de drie virussen in deze studie omvat, en toonde aan dat 1074/1311 (82%) van deze leest kon worden kruis-uitgelijnd met meer dan een viraal genoom (1.047 leest) of kruis-uitgelijnd met verschillende regio’s binnen hetzelfde genoom (27 leest), wat suggereert dat ze chimaera zijn. Om deze waarneming te bevestigen, hebben we onderzocht de ruwe huidige signalen van een paar kruis-gealligneerd leest in vergelijking met die van de juiste toegewezen leest (figuur 2C). De huidige signalen van een correct toegewezen gelezen omvatten meestal: (i) een open porie signaal van hoge stroom die de tijd dat de sequencing porie verandert van de ene adapter naar de andere, (ii) een kraam signaal, verwijzend naar de periode dat een DNA-sequentie is in de porie, maar nog te bewegen, en (iii) het signaal spoor van DNA-sequencing. Een chimere lezing daarentegen heeft een “stall”-signaal en een enorm “spike”-signaal in het midden van de lezing. Chimerische lezingen kunnen twee verschillende barcode-sequenties aan het begin en het einde hebben, waardoor de toewijzing van een barcode bin wordt verward. Samen tonen deze gegevens twee categorieën van fouten aan die bijdragen tot kruissteekproefcontaminatie in onze dataset: (i) chimerische leest (goed voor ∼80% van alle kruiselings toegewezen leest); (ii) leest met een lage barcode score. Om de kwaliteit van onze uiteindelijke dataset te verbeteren, onderzochten we de impact van verschillende barcode demultiplexing benaderingen om kruislings toegewezen leest te verwijderen (tabel 3). Het filteren van de gelezen die een interne adapter bezitten kan 90% van de kruislings toegewezen gelezen verwijderen en verloor 24% van de totale gelezen. We hebben ook geprobeerd een meer stringente filtering regeling die twee streepjescodes (een elk aan het begin en einde van de lees) vereist om een toewijzing te maken. Deze aanpak verwijderde alle behalve twee kruiselings toegewezen leest, maar verloor 56% van de totale leest.

TABLE 3. Verwijdering van cross-assigned reads en verlies van totale sequencing data door twee filtering benaderingen met behulp van Porechop.
We onderzoeken ook de omvang van potentiële chimerische reads in de sequencing data. Voor CHIKV, DENV en FLU-A afzonderlijke sequencing runs, mapping resultaten tonen aan dat 2,3, 3,0 en 2,7% van de gemapte leest, respectievelijk, aanvullende uitlijning bezitten en uitgelijnd ten minste tweemaal naar hetzelfde genoom (tabel 4). We beschouwen zowel de barcode geclassificeerde en niet-geclassificeerde leest in de multiplex sequencing data. Resultaten tonen aan dat 2,0% van de in kaart gebrachte leest bezitten aanvullende uitlijning en uitgelijnd ten minste tweemaal naar hetzelfde genoom, terwijl 0,052% van de totale leest werden uitgelijnd op ten minste twee verschillende genomen.

TABLE 4. Overzicht van het aantal en percentage niet-chimerische, zelf-chimerische en kruis-chimerische lezingen in elke sequencing run.
Discussie
Het uiteindelijke doel van ons onderzoek is om een nanopore metagenomische sequencing gebaseerde diagnostische assay te ontwikkelen die point-of-care testen voor infectieziekten mogelijk maakt. Multiplex sequencing biedt de mogelijkheid om de schaalbaarheid te verbeteren en de kosten te verlagen, echter, cross sample contaminatie kan leiden tot fouten in de gegevens en onjuiste interpretatie van de resultaten.
In dit experiment, we gepoold schone bibliotheken en uitgevoerd multiplex MinION sequencing om de omvang en de bron van cross-barcode contaminatie te onderzoeken. We identificeerden 0,056% van de totale gelezen waren cross-toegewezen aan de onjuiste barcodebakken, wat vergelijkbaar is met die gerapporteerd voor Illumina sequencing platforms uit verschillende studies (tussen 0,06 en 0,25%) (Nelson et al., 2014; D’Amore et al., 2016; Wright en Vetsigian, 2016). Onze resultaten toonden aan dat chimerische reads de overheersende bron van cross-barcode toewijzingsfouten zijn. Cross-toegewezen chimerische leest in deze dataset kon alleen zijn gevormd tijdens sequencing in plaats van bibliotheek voorbereiding, zoals ze waren volledig afwezig in de sequencing gegevens van individuele bibliotheken, en de enige verdere verwerking stap was om de uiteindelijke sequencing bibliotheken te mengen voorafgaand aan het laden. We veronderstellen dat het huidige algoritme geïmplementeerd in Albacore de korte dissociatie tussen DNA-sequenties die gelijktijdig door de nanopore lopen niet kan herkennen, waardoor meer dan één sequentie in hetzelfde Fast5-bestand wordt samengevoegd.
Chimeric reads werden eerder waargenomen in MinION-sequencinggegevens in White et al. (2017). Door analyses van de MinION sequencing data van drie verschillende interferon amplicons, vonden de auteurs dat 1,7% van de gemapte reads chimera waren. Onze bevindingen voegen toe aan de kennis die ondersteunt dat chimera vaak voorkomen in MinION-sequencinggegevens. Wij identificeerden tussen 2 en 3% van de totale gelezen in drie individuele en een multiplex sequencing gegevens zijn chimera. Onze studie verschilt van eerder werk in de volgende twee aspecten. Ten eerste leveren we direct bewijs dat chimerische leest kan worden gevormd na de bibliotheek voorbereiding en tijdens sequencing, we verder gekoppeld deze chimaera cross-sample verontreiniging in multiplex MinION sequencing zoals hierboven besproken. Aan de andere kant, onze experiment setup heeft beperking in het identificeren van potentiële chimaera gevormd in de bibliotheek voorbereiding, met name tijdens de adaptor ligatie stap in de standaard multiplex sequencing protocol. Ten tweede, onze bevindingen weerspiegelen de huidige status van MinION sequencing omdat we nieuwere en meest representatieve ONT sequencing kit, waaronder ligatie sequencing kit 1D (SQK-LSK108) en natieve barcoding kit 1D (EXP-93 NBD103) gebruikt. De nanopore sequencingtechnologie is volop in ontwikkeling en wordt in alle opzichten verbeterd. Zo zijn bijvoorbeeld een nieuwere DNA ligatie sequencing kit (SQK-LSK109) en een directe RNA sequencing kit (SQK-RNA001) uitgebracht; het basecalling algoritme dat in Albacore en Guppy basecaller is geïmplementeerd, is verbeterd. Al deze veranderingen hebben effect op de mate van chimaera in Nanopore sequencing gegevens en cross-barcode besmetting tijdens multiplex sequencing. De beperking van deze studie was het kleine aantal experimenten, extra werk met behulp van verschillende experiment setups zou toevoegen aan ons begrip van Nanopore multiplex sequencing gegevens. Bovendien is het belangrijk om de bijdragen van potentiële factoren aan cross-barcode vervuiling, die licht zou werpen op de beste praktijk om multiplex sequencing data.
In samenvatting, onze studie aangetoond dat chimere leest zijn de overheersende bron van cross-barcode toewijzing fouten in multiplex MinION sequencing. Het benadrukt de noodzaak van zorgvuldige filtering van multiplex MinION sequencing gegevens vóór downstream-analyse, en de trade-off tussen gevoeligheid en specificiteit die geldt voor de barcode demultiplexing methoden.
Author Contributions
SP, KL, SL, en YX uitgevoerd MinION sequencing. YX analyseerde de gegevens. Alle auteurs ontwierpen de studie, namen deel aan de interpretatie van de resultaten en het schrijven van het manuscript, en lazen en keurden de definitieve versie van dit manuscript goed.
Funding
Dit werk werd ondersteund door NIHR Oxford Biomedical Research Centre.
Conflict of Interest Statement
De auteurs verklaren dat het onderzoek werd uitgevoerd in afwezigheid van enige commerciële of financiële relaties die zouden kunnen worden opgevat als een potentieel belangenconflict.
Acknowledgments
Wij danken Dr. Anthony Marriott (Public Health England) voor het ter beschikking stellen van frettenneusaspiraten.