Wiele z metod wizualizacji i interpretacji danych o ekspresji genów może być stosowanych zarówno w przypadku eksperymentów mikromacierzowych, jak i RNA-seq. Niektóre z najczęściej stosowanych metod omówiono poniżej.
Mapy cieplne i grupowanie
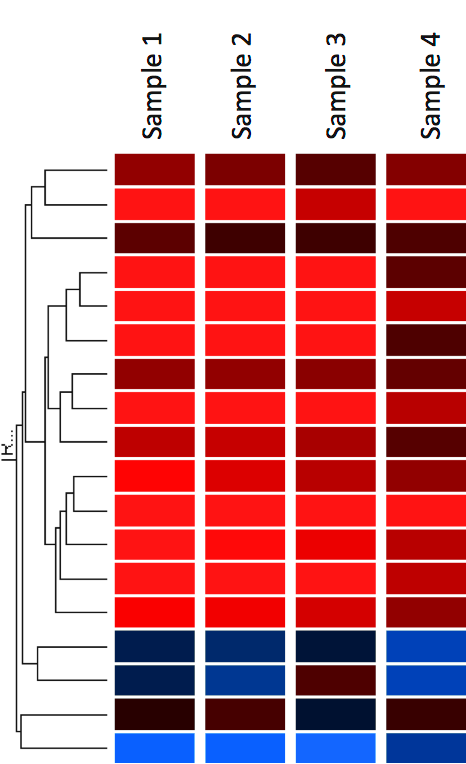
Powszechną metodą wizualizacji danych o ekspresji genów jest wyświetlanie ich w postaci mapy cieplnej (Rysunek 12). Mapa cieplna może być również połączona z metodami klasteryzacji, które grupują geny i/lub próbki razem na podstawie podobieństwa ich wzorca ekspresji genów. Może to być przydatne do identyfikacji genów, które są wspólnie regulowane lub sygnatur biologicznych związanych z określonym stanem (np. chorobą lub stanem środowiskowym) (4).
W mapach cieplnych dane są wyświetlane w siatce, gdzie każdy rząd reprezentuje gen, a każda kolumna reprezentuje próbkę. Kolor i intensywność pól jest używana do reprezentowania zmian (nie wartości bezwzględnych) ekspresji genów. W poniższym przykładzie kolor czerwony reprezentuje geny o podwyższonej ekspresji, a niebieski geny o obniżonej ekspresji. Kolor czarny reprezentuje niezmienioną ekspresję.
Analiza wzbogacania zbioru genów i analiza szlaków
Powszechnym podejściem do interpretacji danych o ekspresji genów jest analiza wzbogacania zbioru genów oparta na funkcjonalnej anotacji genów ulegających różnej ekspresji (Rysunek 13). Jest to przydatne, aby dowiedzieć się, czy różnie wyrażone geny są związane z określonym procesem biologicznym lub funkcją molekularną.
Ontologia genów, zawierająca znormalizowaną adnotację produktów genowych, jest powszechnie używana do tego celu. Działa ona poprzez porównanie częstości występowania poszczególnych adnotacji na liście genów (np. genów różnie wyrażonych) z listą referencyjną (zwykle wszystkie geny na mikromacierzy lub w genomie). W podobny sposób można wzbogacać ścieżki biologiczne dostarczane przez KEGG, Ingenuity, Reactome lub WikiPathways (12,13).
Popularne narzędzia do wzbogacania zbiorów genów i analizy ścieżek obejmują:
- DAVID (darmowe narzędzie online)
- GSEA (darmowe)
- Ingenuity (wymagana licencja)
- Reactome (darmowe)

Analiza sieciowa
Analiza sieciowa jest uzupełnieniem analizy ścieżek i może być wykorzystana do pokazania, jak kluczowe składniki różnych ścieżek wchodzą w interakcje. Może to być przydatne do identyfikacji zdarzeń regulacyjnych, które wpływają na wiele procesów biologicznych i ścieżek (12,13).