- Wprowadzenie
- Materiały i metody
- Przygotowanie próbek
- Przygotowanie bibliotek MinION i sekwencjonowanie
- Analiza genomiczna
- Wyniki
- Dane sekwencjonowania MinION i składanie genomów wirusowych
- Extent and Source of Cross-Sample Contamination
- Dyskusja
- Wkład autorów
- Funding
- Oświadczenie o konflikcie interesów
- Podziękowania
- Materiały uzupełniające
- Przypisy
Wprowadzenie
Sekwencjonowanie metagenomiczne ma potencjał, aby umożliwić bezstronną identyfikację patogenów z próbek klinicznych. Obiecuje służyć jako pojedynczy i uniwersalny test do diagnostyki chorób zakaźnych bezpośrednio z próbek bez potrzeby posiadania wiedzy a priori (Bibby, 2013; Miller i in., 2013; Schlaberg i in., 2017). Oprócz identyfikacji gatunków patogenów, szerokie i głębokie dane sekwencji metagenomicznych mogą dostarczyć informacji istotnych dla określenia leczenia i rokowania, wykrywania ognisk i śledzenia epidemiologii zakażeń (Greninger i in., 2010; Yang i in., 2011; Qin i in., 2012; Loman i in., 2013). Platformy sekwencjonowania następnej generacji (NGS) mogą generować ogromne ilości danych przy umiarkowanych kosztach, jednak ich zastosowanie w diagnostyce klinicznej i zdrowiu publicznym jest ograniczone przez złożoność, powolność i nakłady inwestycyjne.
MinION to sekwenator genomowy wielkości dłoni, działający w czasie rzeczywistym, z pojedynczymi cząsteczkami, opracowany przez Oxford Nanopore Technologies (ONT). Niewielkie rozmiary MinION i jego charakter w czasie rzeczywistym mogłyby ułatwić zastosowanie sekwencjonowania metagenomicznego w badaniach chorób zakaźnych w punktach opieki, jak wykazało kilka badań proof-of-concept, w tym identyfikacja wirusa Chikungunya (CHIKV), Ebola (EBOV) i wirusa zapalenia wątroby typu C (HCV) z ludzkich próbek krwi klinicznej bez wzbogacania celu (Greninger i in., 2015), oraz wykrywanie patogenów bakteryjnych z próbek moczu (Schmidt i in., 2016) i próbek układu oddechowego, bez konieczności wcześniejszej hodowli (Pendleton i in., 2017).
Wydajność danych MinION znacznie wzrosła od czasu jego premiery w 2015 roku, przy czym każda zużywalna komórka przepływowa generuje obecnie do 10-20 Gb danych sekwencji DNA. Pozwala to użytkownikom na bardziej efektywne wykorzystanie komórki przepływowej (i obniżenie kosztów) poprzez multipleksowanie kilku próbek w pojedynczym przebiegu sekwencjonowania. Firma ONT opracowała zestawy kodów kreskowych bez PCR, które umożliwiają multipleksowanie do 12 próbek.
Detekcja wirusa grypy A w wielu próbkach układu oddechowego może być jednym z zastosowań diagnostycznych multipleksowanego testu sekwencjonowania MinION. Jednakże, podczas sekwencjonowania bezpośrednio z próbek o potencjalnie szerokim zakresie miana wirusa, ważne jest, aby być świadomym możliwości zanieczyszczenia krzyżowego próbek, zarówno podczas przygotowania biblioteki, jak i bioinformatycznego etapu demultipleksacji kodów kreskowych po sekwencjonowaniu. Przedstawiamy tu unikalny zestaw danych sekwencjonowania MinION i wyniki badań nad zakresem i źródłem krzyżowego zanieczyszczenia kodów kreskowych w sekwencjonowaniu multipleksowym.
Materiały i metody
Użyliśmy próbki popłuczyn nosowych fretek zakażonych wirusem grypy A jako próbki wzorcowej, a także wprowadziliśmy do dwóch alikwotów negatywnych próbek popłuczyn nosowych niezakażonych fretek (istniejące wcześniej niewykorzystane zapasy z niepowiązanego badania) osobno wirusy dengi i chikungunya. Żaden z tych wirusów nie ma znaczenia dla diagnostyki klinicznej w próbkach układu oddechowego, ale tutaj działają jako wyraźne, odrębne markery do oceny zanieczyszczenia krzyżowego próbek. Biblioteki sekwencjonujące dla każdej próbki były przygotowywane równolegle, wraz z negatywną kontrolą popłuczyn nosowych, kodowane paskowo i sekwencjonowane indywidualnie. Następnie połączyliśmy podwielokrotność bibliotek sekwencjonowania i przeprowadziliśmy sekwencjonowanie multipleksowe MinION. Odczyty z czterech pojedynczych badań (określanych jako „CHIKV”, „DENV”, „FLU-A” i „Negative”) oraz badania multipleksowego (określanego jako „Multiplexed”) zostały następnie przeanalizowane w celu zbadania zakresu i źródła kontaminacji krzyżowej próbek.
Przygotowanie próbek
Koncesja projektu została zweryfikowana przez lokalną AWERB (Animal Welfare and Ethics Review Board), a następnie przyznana przez Home Office. RNA ekstrahowano przy użyciu zestawu QIAamp viral RNA (Qiagen) zgodnie z instrukcjami producenta z popłuczyn nosowych fretek zawierających wirus grypy A (H1N1) (A/California/04/2009) oraz z puli negatywnych próbek popłuczyn nosowych. Kwantyle ekstraktu z próbek ujemnych nasączono wirusem dengi (DENV) (szczep TC861HA, GenBank: MF576311) lub CHIKV (szczep S27, GenBank: MF580946.1) z Krajowej kolekcji wirusów patogennych1. Próbki poddano działaniu DNazy przy użyciu TURBO DNase (Thermo Fisher Scientific, Waltham, MA, Stany Zjednoczone) i oczyszczono przy użyciu zestawu RNA Clean & ConcentratorTM-5 (Zymo Research). cDNA przygotowano i amplifikowano przy użyciu metod Sequence-Independent-Single-Primer-Amplification (Greninger i in., 2015) zmodyfikowanych zgodnie z wcześniejszym opisem (Atkinson i in., 2016). Amplifikowane cDNA zostało określone ilościowo przy użyciu Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA, Stany Zjednoczone), a 1 μg zostało użyte jako dane wejściowe do każdego przygotowania biblioteki MinION, z wyjątkiem kontroli negatywnej, gdzie użyto całej próbki (32 ng).
Przygotowanie bibliotek MinION i sekwencjonowanie
Zestaw do sekwencjonowania ligacyjnego 1D (SQK-LSK108) i zestaw do kodowania natywnego 1D (EXP-NBD103) były używane zgodnie ze standardowymi protokołami ONT, z wyjątkiem tego, że tylko jeden kod kreskowy był zawarty w każdym z czterech preparatów bibliotecznych. Każda biblioteka była uruchamiana na indywidualnej komórce przepływowej, a piąta biblioteka zbiorcza była tworzona przez połączenie czterech indywidualnie kodowanych bibliotek. Biblioteki były sekwencjonowane na komórkach przepływowych R9.4. Projekt badania przedstawiono na rysunku 1.
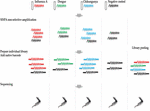
FIGURACJA 1. Przegląd projektu badania. RNA ekstrahowano z czterech próbek, w tym z próbki popłuczyn nosowych fretek zakażonych wirusem grypy A, dwóch negatywnych próbek popłuczyn nosowych fretek zakażonych wirusami dengi i chikungunya oraz negatywnej kontroli popłuczyn nosowych fretek. cDNA przygotowywano i amplifikowano przy użyciu metody Sequence-Independent-Single-Primer-Amplification. Biblioteki sekwencjonujące dla każdej próbki były przygotowywane równolegle, znakowane kodami kreskowymi i sekwencjonowane na pojedynczych komórkach przepływowych. Wykonano również sekwencjonowanie multipleksowe poprzez łączenie czterech indywidualnych bibliotek. Odczyty z czterech pojedynczych przebiegów i przebiegu multipleksowego analizowano w celu oceny zakresu i źródła krzyżowego zanieczyszczenia kodami kreskowymi w sekwencjonowaniu multipleksowym.
Analiza genomiczna
Odczyty poddano basecalled przy użyciu Albacore v2.1.7 (ONT) z demultipleksacją kodów kreskowych. Odczyty z każdego przebiegu sekwencjonowania mapowano do sekwencji genomowych każdego wirusa przy użyciu Minimap2 (Li, 2018). Liczba odczytów zmapowanych do referencji została policzona przy użyciu Pysam2. Montaż de novo wykonano przy użyciu Canu v1.7 (Koren i in., 2017), a wynikowy draft genomu wypolerowano przy użyciu Nanopolish (Mongan i in., 2015) z danymi na poziomie sygnału.
Aby umożliwić rygorystyczne demultipleksowanie kodów kreskowych danych z multipleksowego sekwencjonowania MinION, przeprowadziliśmy dwie rundy analiz przy użyciu Porechop (v0.2.23). Obecność sekwencji adaptera w środku odczytu jest sygnaturą chimery. Użyliśmy Porechop do zbadania każdego odczytu i te, które miały środkowy region dzielący >75% identyczności z sekwencją adaptera zostały zidentyfikowane jako odczyty chimeryczne. W Porechop ustawiliśmy opcję „-middle_threshold” i wybraliśmy próg 75. W drugiej rundzie użyliśmy Porechop do wyszukania sekwencji kodu kreskowego zarówno na początku, jak i na końcu odczytu; odczyty zostały przypisane tylko wtedy, gdy na dwóch końcach znaleziono ten sam kod kreskowy. W Porechopie ustawiliśmy opcję „-require_two_barcodes” i ustawiliśmy próg wyniku dla kodu kreskowego na 70. Aby znaleźć potencjalne sygnatury chimerycznych odczytów, zbadaliśmy sygnały prądowe odczytów zapisane w pliku FAST5 przez sekwenator MinION. Sygnały prądowe zostały wyekstrahowane przy użyciu ONT fast5 API4 i wykreślone przy użyciu ggplot2 zaimplementowanego w R5 w celu porównania odczytów chimerycznych i niechimerycznych.
Wyniki
Dane sekwencjonowania MinION i składanie genomów wirusowych
Wydajność każdego przebiegu sekwencjonowania MinION różniła się ze względu na różnice w czasie pracy. Maksymalna liczba ∼ 2,4 M odczytów została osiągnięta w badaniu sekwencjonowania multipleksowego i indywidualnego badania CHIKV, z powodu dłuższego czasu pracy (Tabela Uzupełniająca S1). Odczyty z wirusa spiked stanowiły 96% danych w indywidualnych przebiegach sekwencjonowania CHIKV i DENV, i 78% dla próbki FLU-A (Tabela 1). Odsetek wirusowych odczytów w obrębie każdej próbki z kodem paskowym w danych sekwencjonowania multipleksowego jest zbliżony do tego w danych z indywidualnie badanych próbek (Tabela 2). Każdy genom wirusowy miał bardzo wysoką (>8,000) średnią głębokość pokrycia w danych sekwencjonowania indywidualnego i multipleksowego, a montaż de novo był w stanie odzyskać prawie kompletne genomy dla wszystkich trzech wirusów z 99.9% identyczności w stosunku do referencji GenBank.

TABELA 1. Podsumowanie wyników mapowania i montażu de novo dla danych z sekwencjonowania MinION poszczególnych bibliotek.

TABELA 2. Podsumowanie wyników mapowania i montażu de novo dla danych z multipleksowego sekwencjonowania MinION.
Extent and Source of Cross-Sample Contamination
Każda próbka była oznaczona kodem kreskowym i sekwencjonowana zarówno indywidualnie, jak i multipleksowo, co pozwoliło nam zbadać wydajność demultipleksowania kodu kreskowego Albacore. W danych z indywidualnie sekwencjonowanych próbek spodziewamy się obecności tylko jednego natywnego kodu kreskowego. W przypadku CHIKV (kod kreskowy NB01), DENV (NB09) i FLU-A (NB10), stwierdziliśmy, że odpowiednio 86, 109 i 17 odczytów zostało przypisanych do koszy kodów kreskowych, które nie powinny być obecne w bibliotece (co stanowi 0,0036, 0,0129 i 0,001% wszystkich odczytów). W danych z sekwencjonowania multipleksowego 41 odczytów (0,0016%) przypisano do kodów kreskowych nieuwzględnionych w eksperymentach (tj. kodów kreskowych innych niż NB01, NB05, NB09 lub NB10). Zdefiniowaliśmy je jako błędnie przypisane odczyty (Figura 2A).
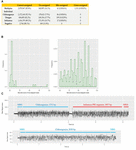
FIGURACJA 2. (A) podsumowanie liczby i odsetka odczytów prawidłowo przypisanych, nieprzypisanych, błędnie przypisanych i krzyżowo przypisanych w każdym przebiegu sekwencjonowania. Nieprzypisane odnoszą się do odczytów, których Albacore nie może przypisać do żadnego z bloków z powodu wyniku kodu kreskowego mniejszego niż 60, błędnie przypisane odnoszą się do odczytów, które zostały przypisane do bloków kodu kreskowego nieobjętych tym eksperymentem, a krzyżowo przypisane odnoszą się do odczytów, które zostały przypisane do nieprawidłowych bloków kodu kreskowego; (B) rozkład wyników kodu kreskowego zgłoszonych przez Albacore dla błędnie przypisanych i krzyżowo przypisanych odczytów w danych sekwencjonowania multipleksowego; (C) porównanie surowego sygnału odczytu chimerycznego i prawidłowo przypisanego. Sygnał odczytu chimerycznego posiada sygnał przeciągnięcia i duży sygnał spike w środku odczytu.
Aby zbadać potencjalne zanieczyszczenie laboratoryjne podczas przygotowywania biblioteki sekwencjonowania, zmapowaliśmy wszystkie odczyty z każdego pojedynczego przebiegu względem sekwencji genomowych wszystkich trzech wirusów. Nie znaleziono żadnego odczytu pochodzącego z genomu przygotowanego w innej bibliotece, co sugeruje brak zanieczyszczenia in vitro. Biblioteka do sekwencjonowania multipleksowego została przygotowana przez połączenie poszczególnych, niezanieczyszczonych bibliotek po ligacji zarówno kodu kreskowego, jak i adaptera. Jednakże, wyniki mapowania wykazały, że 1,311 (0.0543%) odczytów zostało zmapowanych do niewłaściwego genomu docelowego, co sugeruje, że zostały one przypisane krzyżowo do niewłaściwych koszy kodów kreskowych (zwanych później „odczytami przypisanymi krzyżowo”), pomimo faktu, że multipleksowana biblioteka sekwencyjna została połączona z indywidualnymi bibliotekami nie wykazała w ogóle odczytów przypisanych krzyżowo. Postawiliśmy hipotezę, że błędnie przypisane i krzyżowo przypisane odczyty były spowodowane niską punktacją kodu kreskowego i zbadaliśmy punktację kodu kreskowego tych odczytów. Większość błędnie przypisanych odczytów miała wynik kodu kreskowego <70, jednak odczyty przypisane krzyżowo miały bardziej zróżnicowane wyniki, od 60 do prawie 100 (Rysunek 2B). Wynik ten sugerował, że błędnie przypisane i krzyżowo przypisane odczyty pochodzą z różnych źródeł. Sprawdziliśmy krzyżowo przypisane odczyty do małej bazy danych zawierającej sekwencje genomowe trzech wirusów objętych tym badaniem i wykazaliśmy, że 1074/1311 (82%) z tych odczytów może być krzyżowo wyrównane do więcej niż jednego genomu wirusa (1047 odczytów) lub krzyżowo wyrównane do różnych regionów w obrębie tego samego genomu (27 odczytów), co sugeruje, że są to chimery. Aby potwierdzić tę obserwację, zbadaliśmy surowe sygnały prądowe kilku krzyżowo przypisanych odczytów w porównaniu z sygnałami prawidłowo przypisanych odczytów (Figura 2C). Bieżące sygnały prawidłowo przypisanego odczytu zwykle zawierają: (i) sygnał otwartego porów o wysokim prądzie reprezentujący czas, w którym pory sekwencjonowania zmieniają się z jednego adaptera na inny, (ii) sygnał przeciągnięcia, odnoszący się do okresu czasu, w którym sekwencja DNA znajduje się w porach, ale jeszcze się nie porusza, oraz (iii) ślad sygnału sekwencjonowania DNA. W przeciwieństwie do tego, odczyt chimeryczny posiada sygnał przeciągnięcia i ogromny sygnał spike w środku odczytu. Odczyty chimeryczne mogą posiadać dwie różne sekwencje kodu kreskowego na początku i na końcu, co powoduje, że przypisanie bin kodu kreskowego jest mylące. Dane te wskazują na dwie kategorie błędów, które przyczyniają się do kontaminacji krzyżowej próbek w naszym zbiorze danych: (i) odczyty chimeryczne (stanowią ∼80% wszystkich przypisanych krzyżowo odczytów); (ii) odczyty z niskim wynikiem kodu kreskowego. W celu poprawy jakości naszego ostatecznego zbioru danych, zbadaliśmy wpływ różnych metod demultipleksacji kodów kreskowych w celu usunięcia krzyżowo przypisanych odczytów (Tabela 3). Filtrowanie odczytów, które posiadają wewnętrzny adapter może usunąć 90% krzyżowo przypisanych odczytów i stracić 24% wszystkich odczytów. Wypróbowaliśmy również bardziej rygorystyczny schemat filtrowania, który wymagał dwóch kodów kreskowych (po jednym na początku i na końcu odczytu), aby dokonać przypisania. To podejście usunęło wszystkie odczyty poza dwoma przypisanymi krzyżowo, ale straciło 56% wszystkich odczytów.

TABELA 3. Usuwanie krzyżowo przypisanych odczytów i utrata całkowitych danych sekwencjonowania przez dwa podejścia filtrujące przy użyciu Porechop.
Badamy również zakres potencjalnych chimerycznych odczytów w danych sekwencjonowania. Dla pojedynczych przebiegów sekwencjonowania CHIKV, DENV i FLU-A, wyniki mapowania pokazują, że odpowiednio 2,3, 3,0 i 2,7% zmapowanych odczytów posiada uzupełniające wyrównanie i wyrównuje się co najmniej dwukrotnie do tego samego genomu (Tabela 4). W danych pochodzących z sekwencjonowania multipleksowego uwzględniliśmy zarówno odczyty sklasyfikowane, jak i niesklasyfikowane za pomocą kodu kreskowego. Wyniki pokazują, że 2,0% zmapowanych odczytów posiada wyrównanie uzupełniające i wyrównuje się co najmniej dwukrotnie do tego samego genomu, podczas gdy 0,052% wszystkich odczytów zostało wyrównanych do co najmniej dwóch różnych genomów.

TABELA 4. Zestawienie liczby i odsetka odczytów niechimerycznych, samo-chimerycznych i krzyżowych w każdym przebiegu sekwencjonowania.
Dyskusja
Najważniejszym celem naszych badań jest opracowanie testu diagnostycznego opartego na metagenomicznym sekwencjonowaniu nanoporowym, który umożliwiłby testowanie chorób zakaźnych w punktach opieki. Multipleksowe sekwencjonowanie oferuje możliwość poprawy skalowalności i obniżenia kosztów, jednak zanieczyszczenie krzyżowe próbek może prowadzić do błędów w danych i fałszywej interpretacji wyników.
W tym eksperymencie połączyliśmy czyste biblioteki i przeprowadziliśmy multipleksowe sekwencjonowanie MinION w celu zbadania zakresu i źródła zanieczyszczenia kodem krzyżowym. Zidentyfikowaliśmy 0,056% całkowitych odczytów zostało przypisanych krzyżowo do nieprawidłowych koszy kodów kreskowych, co jest porównywalne z danymi zgłoszonymi dla platform sekwencjonowania Illumina z różnych badań (między 0,06 a 0,25%) (Nelson i in., 2014; D’Amore i in., 2016; Wright i Vetsigian, 2016). Nasze wyniki pokazały, że chimeryczne odczyty są dominującym źródłem błędów przypisania kodów krzyżowych. Przypisane krzyżowo odczyty chimeryczne w tym zbiorze danych mogły powstać tylko podczas sekwencjonowania, a nie przygotowania bibliotek, ponieważ były one całkowicie nieobecne w danych sekwencjonowania poszczególnych bibliotek, a jedynym dalszym etapem przetwarzania było mieszanie końcowych bibliotek sekwencjonujących przed załadowaniem. Stawiamy hipotezę, że obecny algorytm zaimplementowany w Albacore nie jest w stanie rozpoznać krótkiej dysocjacji między sekwencjami DNA, które przebiegają równolegle przez nanopor, konkatenując w ten sposób więcej niż jedną sekwencję w tym samym pliku Fast5.
Chimeryczne odczyty zaobserwowano w danych sekwencjonowania MinION wcześniej w White et al. (2017). Poprzez analizy danych sekwencjonowania MinION trzech różnych amplikonów interferonu, autorzy stwierdzili, że 1,7% zmapowanych odczytów stanowiły chimery. Nasze wyniki dodają do wiedzy wspierającej, że chimery są powszechne w danych sekwencjonowania MinION. Zidentyfikowaliśmy, że od 2 do 3% wszystkich odczytów w trzech pojedynczych i jednym sekwencjonowaniu multipleksowym to chimery. Nasze badania różnią się od poprzednich prac w następujących dwóch aspektach. Po pierwsze, dostarczamy bezpośrednich dowodów na to, że chimeryczne odczyty mogą powstawać po przygotowaniu biblioteki i podczas sekwencjonowania; następnie powiązaliśmy te chimery z zanieczyszczeniem krzyżowym próbek w multipleksowym sekwencjonowaniu MinION, jak omówiono powyżej. Z drugiej strony, nasza konfiguracja eksperymentu ma ograniczenia w identyfikacji potencjalnych chimer powstałych podczas przygotowania biblioteki, w szczególności podczas etapu ligacji adaptora w standardowym protokole sekwencjonowania multipleksowego. Po drugie, nasze wyniki odzwierciedlają obecny status sekwencjonowania MinION, ponieważ użyliśmy nowszego i najbardziej reprezentatywnego zestawu do sekwencjonowania ONT, w tym zestawu do sekwencjonowania z ligacją 1D (SQK-LSK108) i zestawu do natywnego kodowania paskowego 1D (EXP-93 NBD103). Technologia sekwencjonowania nanoporowego jest w trakcie szybkiego rozwoju i udoskonalania we wszystkich aspektach. Na przykład, wprowadzono nowszy zestaw do sekwencjonowania z ligacją DNA (SQK-LSK109) i zestaw do bezpośredniego sekwencjonowania RNA (SQK-RNA001); unowocześniono algorytm basecallingu zaimplementowany w basecallerach Albacore i Guppy. Wszystkie te zmiany mają wpływ na zakres chimery w danych sekwencjonowania Nanopore oraz zanieczyszczenie cross-barcode podczas sekwencjonowania multipleksowego. Ograniczeniem tego badania była mała liczba eksperymentów, dodatkowe prace z wykorzystaniem różnych konfiguracji eksperymentów przyczyniłyby się do lepszego zrozumienia danych sekwencjonowania multipleksowego Nanopore. Ponadto, ważne jest zbadanie udziału potencjalnych czynników w zanieczyszczeniu kodem kreskowym, co rzuciłoby światło na najlepsze praktyki analizy danych sekwencjonowania multipleksowego.
Podsumowując, nasze badanie wykazało, że chimeryczne odczyty są dominującym źródłem błędów przypisania kodu kreskowego w sekwencjonowaniu multipleksowym MinION. Podkreśla to potrzebę starannego filtrowania danych multipleksowego sekwencjonowania MinION przed dalszą analizą oraz kompromis pomiędzy czułością i specyficznością, który dotyczy metod demultipleksacji kodów kreskowych.
Wkład autorów
SP, KL, SL i YX przeprowadzili sekwencjonowanie MinION. YX dokonał analizy danych. Wszyscy autorzy zaprojektowali badanie, uczestniczyli w interpretacji wyników i pisaniu manuskryptu oraz przeczytali i zatwierdzili ostateczną wersję tego manuskryptu.
Funding
Ta praca była wspierana przez NIHR Oxford Biomedical Research Centre.
Oświadczenie o konflikcie interesów
Autorzy oświadczają, że badania zostały przeprowadzone przy braku jakichkolwiek komercyjnych lub finansowych powiązań, które mogłyby być interpretowane jako potencjalny konflikt interesów.
Podziękowania
Chcielibyśmy podziękować Dr. Anthony Marriott (Public Health England) za dostarczenie aspiratów nosowych fretek.
Materiały uzupełniające
Przypisy
.