INTRODUÇÃO
Doenças pulmonares intersticiais formam um grupo extremamente heterogéneo de doenças que têm em comum a existência de um componente inflamatório do parênquima pulmonar que pode eventualmente evoluir para fibrose e que causam, do ponto de vista funcional, uma alteração restritiva que pode eventualmente levar à insuficiência respiratória.
P Embora se trate de doenças cujo diagnóstico e tratamento é realizado no campo especializado da pneumologia, o tratamento é fundamentalmente seguido em regime ambulatório, e dado que este pode e muitas vezes é pontuado por várias complicações, é importante que o médico da atenção primária esteja atento às directrizes actuais para o tratamento destas doenças, pois em muitas ocasiões será consultado por pacientes com este tipo de problemas, quer devido a complicações respiratórias, quer devido a outros tipos de complicações.
O objetivo desta revisão é atualizar as modalidades terapêuticas que podem ser oferecidas hoje a esses pacientes. Acreditamos que é da maior importância não só para o médico da atenção primária ter um conhecimento atualizado sobre este assunto, mas também para colaborar com o pneumologista, a fim de obter uma boa adesão ao tratamento do paciente e detecção precoce e tratamento de quaisquer complicações que possam surgir.
Dentre as doenças intersticiais, a mais relevante devido à sua freqüência e gravidade é a fibrose pulmonar idiopática (FPI). Por outro lado, o seu tratamento é o modelo em que se baseia o tratamento de quase todas estas doenças, com excepção daquelas em que se pode encontrar um agente etiológico conhecido, uma vez que nelas a eliminação deste agente será a base fundamental da terapia. Portanto, vamos nos concentrar em uma revisão das diretrizes atuais para a gestão do IPF. Vamos primeiro rever brevemente as suas características clínicas e depois desenvolver em detalhe a gestão actual da IPF.
DESCRIÇÃO CLÍNICA
IPF representa provavelmente o protótipo mais agressivo das pneumopatias intersticiais difusas. Sua prevalência tem sido estimada por alguns autores em 5 casos por 100.000 habitantes1, embora na realidade seja desconhecida. Sua patogênese está relacionada a alterações nas complexas vias do sistema imunológico2,3. Por esta razão, a maioria dos medicamentos utilizados são imunossupressores.
A apresentação clínica mais comum é dispneia de início insidioso acompanhada de tosse seca, que pode ser o sintoma mais incómodo para o paciente devido à sua natureza paroxística e ofegante. Geralmente, a dispneia é, inicialmente, pouco marcante e um alto índice de suspeita é necessário para um diagnóstico precoce. Ocasionalmente, o curso é rápido desde o início e leva, em poucos meses, a uma insuficiência respiratória irreversível, como era característico das descrições iniciais de Hamman e Rich. No entanto, mais comumente, a condição já está presente há meses quando o paciente faz sua primeira consulta.
Ao exame físico, crepitações finas e secas (velcro rales) são muito características e podem ser ouvidas em mais de 80% dos casos. Em casos avançados, pode ser observada dispneia com leve esforço ou mesmo cianose labial. Acropaquias podem ser vistas em cerca de 30-50% dos casos. A suspeita de IPF é reforçada quando sinais como padrão reticular, áreas de infiltração de vidro moído e diminuição do volume pulmonar aparecem na radiografia do tórax e na tomografia computadorizada. Finalmente, o exame funcional confirmará a existência de um padrão restritivo e a análise dos gases do sangue arterial confirmará a existência de hipoxemia. Em casos duvidosos, a demonstração de hipoxemia ao esforço é de grande valor.
A confirmação definitiva do diagnóstico só pode ser feita por biópsia pulmonar. Contudo, a realidade é que, mesmo em muitas das séries publicadas, predominam casos sem diagnóstico histológico. Recentemente, a American Thoracic Society (ATS) propôs que a existência de todos esses critérios significa que, na ausência de exposição a drogas, elementos ambientais ou doença do tecido conjuntivo, o diagnóstico de IPF pode muito provavelmente ser feito na ausência de biópsia pulmonar4 nos casos em que a biópsia pulmonar seria um risco inaceitável.
Pneumonia intersticial oral (PIU) é o padrão histopatológico que identifica esses pacientes4. Aqueles com padrões de pneumonia intersticial desquamatória, doença intersticial associada à bronquiolite respiratória, pneumonia intersticial inespecífica, pneumonia intersticial linfóide, pneumonia intersticial aguda e bronquiolite obliterante com pneumonia organizadora são considerados entidades diferentes.
Após o diagnóstico e a gravidade clínica estarem estabelecidos, a maioria dos pacientes necessita de tratamento por se tratar de uma doença fatal sem remissão espontânea. A resposta ao tratamento é fraca. A sobrevivência em cinco anos é de 50%5,2,6-8. O tratamento requer cerca de 3-6 meses antes que a eficácia seja estabelecida. Os relatos sobre o tratamento destas doenças são escassos: ensaios terapêuticos descontrolados que envolvem, na sua maioria, um pequeno número de pacientes9,
Apesar da inevitável progressão insidiosa e irreversível da doença, o niilismo terapêutico não se justifica. Existem exemplos de fibrose não tratada com progressão lenta que levam a uma fibrose irreversível sem resposta ao tratamento ou desenvolvendo uma fase rapidamente progressiva. Todos provavelmente requerem tratamento no momento da apresentação. No entanto, os pacientes com contra-indicações de tratamento ou aqueles sem evidência de comprometimento funcional podem seguir um programa de acompanhamento em intervalos de 3-6 meses. O manejo de pacientes idosos com histologia de predomínio de fibrose e comprometimento grave da função pulmonar também é difícil. Estes pacientes recebem um tratamento curto de 3-6 meses para determinar a reversibilidade. Poucos terão uma resposta positiva.
É muitas vezes difícil determinar se o tratamento produz o efeito desejado. A melhoria subjectiva ocorre frequentemente (em até 70% dos pacientes). A melhoria objetiva ocorre em 20-30%. Os parâmetros mais utilizados para definir a melhoria funcional (objectiva) são um aumento da capacidade pulmonar total (TLC) ou da capacidade vital (VC), um aumento da capacidade de difusão e uma redução ou normalização do declínio da SaO2 durante o exercício4,10-12.
TREATAMENTO
Corticosteróides
Corticosteróides são a base do tratamento para IPF, embora seu sucesso seja bastante ruim: uma resposta favorável ocorre em um terço dos pacientes. A razão para o seu uso é suprimir a alveolite crónica. Pensa-se que funcionam suprimindo a migração de neutrófilos e linfócitos no pulmão, diminuindo a concentração de imunocomplexos, alterando a função dos macrófagos alveolares e a adesão dos neutrófilos às superfícies endoteliais.
Não está claro porque alguns pacientes respondem e outros não. Pode estar relacionado com a presença de receptores de superfície nas células parenquimatosas pulmonares. Uma evolução dos sintomas a curto prazo, um leve comprometimento funcional e radiológico, sexo feminino e idade mais jovem dos pacientes parecem estar associados a um melhor resultado.
No entanto, apesar de ser o tratamento mais utilizado, seu desempenho a longo prazo não foi confirmado por nenhum estudo prospectivo, mono-cego e placebo-controlado.
Izumi et al analisaram a sobrevida de 222 pacientes e constataram que, aos 10 anos, não houve diferença na sobrevida entre aqueles que receberam esteróides em comparação aos pacientes não tratados13. No entanto, quando um paciente com IPF apresenta função pulmonar comprometida ou sintomas que interferem com a vida normal, é razoável iniciar uma tentativa de terapia com corticosteroides. De facto, a melhoria espontânea da doença é muito rara, e se for decidido adiar o início do tratamento deve ser à custa de uma monitorização próxima do doente para que o tratamento possa ser iniciado assim que a deterioração seja evidente.
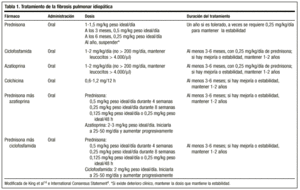
O tratamento clássico começa com prednisona 1,5-2 mg/kg de peso ideal/dia como dose única de manhã, para não exceder uma dose de 100 mg. Após 2-3 meses, o paciente é reavaliado com radiografia e estudo funcional. Se o paciente responder, a prednisona é cónica (1-2 mg/semana) a uma dose de manutenção de 0,5 mg/kg de peso/dia ideal, que pode ser reduzida posteriormente. Se não houver resposta, uma droga imunossupressora é adicionada. Em geral, é improvável que o tratamento possa ser interrompido, e as doses de 0,25 mg/kg de peso ideal/dia são normalmente mantidas.
Recentemente, as Sociedades Torácica Americana e Européia, a fim de evitar os efeitos colaterais deste medicamento e obter melhores taxas de sobrevivência, chegaram a um consenso para iniciar o tratamento com uma combinação de prednisona ou outro corticosteróide em doses equivalentes e um imunossupressor (azatioprina ou ciclofosfamida)4: prednisona 0,5 mg/kg de peso ideal/dia durante 4 semanas; 0,25 mg/kg de peso ideal/dia durante 8 semanas, e depois afunilando para 0,125 mg/kg de peso ideal/dia ou 0,25 mg/kg de peso ideal/48 h.
Não há orientações sobre a duração do tratamento; o tempo mínimo é de um ano. É possível que após meses ou anos a doença possa reativar, caso em que o tratamento com os mesmos agentes deve ser iniciado. Em qualquer caso, a resposta ao tratamento deve ser sempre avaliada em termos de melhoria objetiva, e a possibilidade de manter o paciente cronicamente em uma dose que garanta estabilidade deve ser reservada para os casos em que há melhora com corticosteróides, deterioração no afilamento e nova melhora no aumento da dose novamente. A dose necessária para cada paciente só pode ser determinada caso a caso, dependendo da resposta às alterações na dose. Muitos autores consideram necessário um tratamento vitalício.14
Quando a doença é inicialmente grave, foram propostos corticosteróides de alta dose. Keogh et al têm utilizado corticosteróides parenterais em doses elevadas intermitentes em pacientes com doença grave: metilprednisolona intravenosa, 2 g uma vez por semana, mais prednisona oral, 0,25 mg/kg/dia. A lógica é que eles têm um efeito mais pronunciado na alveolite neutrófila (associada a um mau prognóstico) do que os esteróides de baixa dosagem15. No entanto, este trabalho sofre de algumas deficiências, como o curto seguimento de 6 meses, o pequeno número de pacientes e as doses de prednisona inferiores ao padrão9. O tratamento inicial com doses elevadas de corticosteróides intravenosos (metilprednisolona 250 mg/6 h) tem sido utilizado num esforço para controlar a doença o mais cedo possível quando esta é rapidamente progressiva. Não há estudos controlados para determinar se esses métodos são eficazes; entretanto, doses mais altas parecem ter maior eficácia, embora os efeitos colaterais também sejam maiores.14.
AgENTES CITOTOXICOS
Têm sido classicamente utilizados como alternativa aos corticosteróides quando eram ineficazes, e também quando seus efeitos colaterais impedem sua posterior administração. De acordo com o espírito eminentemente prático deste artigo, vamos rever os agentes que se revelaram úteis na prática clínica, embora, como no caso dos corticosteróides, nos faltem séries longas, prospectivas e aleatórias que nos permitam saber exatamente qual é o seu papel exato no tratamento da IPF.
Azatioprina
O mecanismo de ação do ácido desoxirribonucleico é baseado na substituição de purinas na síntese do ácido desoxirribonucleico, inibindo assim a deaminase adenina e inativando os linfócitos. Outros efeitos incluem a supressão da atividade natural das células assassinas, produção de anticorpos e citotoxicidade celular anti-corpo.
A dose recomendada é de 2 mg/kg/dia, para não exceder 200 mg/dia, por um período de 3-6 meses. A administração é iniciada com uma dose de 50 mg/dia e, se bem tolerada, a dose é aumentada em intervalos de 1 a 2 semanas até que a dose planejada seja alcançada.
Os principais efeitos colaterais são hematológicos: leucopenia, anemia, trombocitopenia, aplasia de eritrócitos e pancitopenia. É necessário monitorar a contagem de leucócitos, e se ela cair abaixo de 4.000/μl, a azatioprina deve ser descontinuada até que esse número aumente novamente. Além disso, pode ocorrer desconforto gastrointestinal, como náuseas e vómitos, úlcera péptica, diarreia e ligeira elevação das enzimas hepáticas. Em geral, em até 20-30% dos casos, os efeitos colaterais requerem a retirada da azatioprina.
Raghu et al. compararam a eficácia dos glicocorticoides de alta dose versus azatioprina em combinação com glucocorticoides de baixa dose. O uso desta droga foi acompanhado por uma modesta melhora nos parâmetros da função respiratória e aumento da sobrevida17,
Ciclofosfamida
É um agente alquilante. O seu modo de acção é a depleção linfocitária, suprimindo assim o funcionamento dos linfócitos. A dose recomendada é de 2 mg/kg/dia, oral, geralmente considerada com 0,25 mg/kg/dia de prednisona oral, não devendo exceder 200 mg/dia durante pelo menos 3 meses e até 9-12 meses.
O efeito colateral mais importante é a leucopenia. Deve ser mantida uma contagem total de leucócitos superior a 3.000/μl ou uma contagem de neutrófilos superior a 1.500/μl. Outras complicações incluem trombocitopenia, hematúria secundária a cistite hemorrágica, anorexia, náuseas e vômitos, supressão de medula óssea, azoospermia e amenorréia, infecção e malignidade hematológica.
Kolb et al estudaram 18 pacientes com IPF para determinar a eficácia e segurança da terapia com ciclofosfamida pulsada. A ciclofosfamida foi administrada intermitentemente (1-1,3 g/month) juntamente com a prednisolona oral durante um ano. Após este período, a ciclofosfamida foi descontinuada e as doses alcançadas de corticosteróides foram continuadas8 . Eles foram seguidos por um mínimo de 14 meses, monitorando QoL, TLC, FEV1, capacidade de transferência de CO (DLCO), SaO2, assim como sintomas. Um paciente teve de ser descontinuado devido a pneumonias recorrentes; 11 pacientes foram considerados respondedores (5 melhoraram e 6 estabilizaram) e 6 deterioraram. A toxicidade a longo prazo da ciclofosfamida depende de doses cumulativas. As doses intermitentes são tão eficazes e melhor toleradas do que as doses diárias. Os respondentes tinham uma maior percentagem de linfócitos no BAL. Outra descoberta notável é que o QOL inicial dos respondedores foi significativamente melhor devido ao menor tempo de progressão, sugerindo que este tratamento deve ser iniciado mais cedo.
Baughman e Lower tratados 33 pacientes IPF com 1.000-1.800 mg de ciclofosfamida/2 semanas durante 18 meses. Aqueles que sobreviveram 18 meses tiveram uma melhora significativa no FCR que foi mantida durante o ano seguinte. A redução de prednisolona foi possível de 32 para 4 mg/dia16,
Penicilamina
Antifibrosante. Tem sido sugerido que ao interferir na formação de ligações covalentes intra e intermoleculares de fibras de colágeno, inibe o desenvolvimento de fibrose pulmonar. Alguns casos de doenças do tecido conjuntivo com fibrose pulmonar associada foram descritos em que teve alguma eficácia, mas não existem estudos controlados em grandes séries de pacientes, pelo que actualmente não pode ser considerado como um tratamento alternativo válido. Portanto, estudos adicionais são necessários antes que uma recomendação possa ser feita.
Colchicina
Diminui a secreção de colágeno e aumenta a atividade colagenolítica. Também tem um efeito anti-inflamatório. São necessários estudos para determinar mais precisamente o seu papel no tratamento desta doença, mas existem estudos em que lhe foi atribuída uma eficácia semelhante à dos corticosteróides, e os seus efeitos secundários não são graves. Recomenda-se uma dose de 0,6 mg, uma ou duas vezes ao dia, em pacientes refratários aos corticosteróides, sozinhos ou em combinação com agentes imunossupressores. Embora existam autores que a colocam na primeira linha de tratamento, atualmente faltam os dados para estabelecê-la como tal (tabela 1).
TRANSPLANTE PULMONAR
Casos em que a doença progride sem resposta ao tratamento e acaba por causar insuficiência respiratória são, infelizmente, os mais numerosos nesta doença. Atualmente não há perspectivas de tratamento médico para estes pacientes, exceto tratamento paliativo. Neste cenário sombrio, a possibilidade de transplante pulmonar surgiu na década de 1980 e desenvolveu-se ao ponto de ser agora uma opção terapêutica que deve ser considerada como uma ferramenta de uso clínico e já não está limitada ao campo experimental. Sua indicação mais adequada é em pessoas com menos de 65 anos de idade, com a ausência de doença progressiva e irreversível em outros órgãos, bem como infecção ativa não resolvida ou infecção por germes multi-resistentes. A predisposição pessoal e o estado nutricional adequados também são necessários. Os pacientes com um CPT inferior a 60% do esperado ou com uma capacidade de difusão pulmonar inferior a 40% têm uma sobrevida inferior a 2 anos. Neste grau de disfunção eles requerem oxigenoterapia e dessaturação fácil com o mínimo de exercício. O transplante pulmonar deve ser considerado nestes casos, assim que a oxigenoterapia se torne necessária e se verifique a progressão imparável da doença. O tipo de transplante de escolha é o transplante de um pulmão, reservando o transplante de dois pulmões para os casos em que haja dúvidas sobre o comportamento do pulmão residual. Quando o paciente tem menos de 50 anos de idade e insuficiência cardíaca direita, o transplante cardiopulmonar pode ser considerado. Contudo, nesta situação, a doença está muito avançada e é pouco provável que resista à espera18,19. A sobrevida pós-transplante é esperada em 80% no primeiro ano e 50-60% aos 5 anos, o que melhora significativamente o prognóstico para um paciente com IPF que atingiu insuficiência respiratória.
EVALIAÇÃO DA RESPOSTA AO TRATAMENTO
Na ausência de complicações, pode levar até 6 meses até que qualquer resposta ao tratamento seja obtida. Após este tempo, se o paciente tiver melhorado ou se estiver estável, as mesmas doses devem ser mantidas. Se tiverem piorado, devem ser mudados para terapia alternativa (outro agente imunossupressor ou mesmo transplante). Aos 12 meses, deve ser reavaliado da mesma forma e após 18 meses, individualizado de acordo com a resposta. Se recomienda continuar indefinidamente sólo en aquellos pacientes con mejoría continua o estabilización (tabla 2).

Una respuesta favorable se define por dos o más de los siguientes criterios en dos visitas consecutivas:
1. Mejoría de los síntomas, sobre todo en el grado de esfuerzo requerido para que el paciente deba parar cuando hace un esfuerzo o un descenso en la frecuencia o gravedad de la tos.
2. Reducción de las alteraciones radiológicas.
3. Mejoría funcional definida por dos o más de los siguientes parámetros:
Aumento >= 10% en la TLC o CV (o >= 200 ml).
Aumento >= 15% en DLCO o >= 3 ml/min/mmHg.
Mejoría o normalización de la SaO2 o pO2 (aumento >= 4% o >= 4 mmHg) durante un prueba de esfuerzo cardiorrespiratoria.
Una respuesta estable (también considerada favorable) se define por dos o más de los siguientes criterios en dos visitas consecutivas:
1. Cambio de
2. Cambio de
3. Sin cambios en la SaO2 o pO2 (aumento
Un fracaso del tratamiento se define por dos o más de los siguientes criterios en dos visitas consecutivas:
1. Aumento de los síntomas, sobre todo disnea o tos.
2. Aumento de las alteraciones radiológicas, especialmente imágenes en panal o signos de hipertensión pulmonar.
3. Deterioro funcional definido por dos o más de los siguientes parámetros:
Descenso >= 10% en la TLC o CV (o >= 200 ml).
Descenso >= 15% en DLCO o >= 3 ml/min/mmHg.
P>Avaliando SaO2 (= 4%) ou elevação do gradiente alveolar-arterial (AapO2) (= 4 mmHg) em repouso ou durante um teste de esforço cardiorrespiratório.
CONCLUSÕES
Fibrose pulmonar idiopática é uma doença fatal, sem remissão espontânea e com má resposta ao tratamento. Os estudos sobre estes últimos são escassos e envolvem um pequeno número de pacientes. Corticosteróides mantidos por um mínimo de um ano em doses diferentes são o regime mais aceito, isoladamente ou em combinação com outros agentes imunossupressores. Recentemente, a terapia combinada tem sido defendida pelas Sociedades Torácica Americana e Respiratória Europeia4 . Os agentes anti-fibrosantes estão sendo investigados com a intenção de evitar que o processo fibrótico se estabeleça e, portanto, irreversível, já que muitos pacientes são diagnosticados em estágios tardios. Dadas as taxas de sobrevivência e evolução, o transplante pulmonar deve ser considerado assim que os pacientes falham o tratamento e necessitam de oxigenoterapia. Tudo isso sugere a necessidade de estabelecer estudos multicêntricos com a idéia de recrutar um número suficiente de pacientes para avaliar a eficácia dos tratamentos administrados, bem como para identificar a história natural da doença e a incidência e prevalência reais4,20. Por outro lado, recomenda-se identificar e iniciar o tratamento nos estágios iniciais da doença, a fim de obter maiores taxas de sobrevivência.