Na celém světě trpí úplnou ztrátou zraku 36 milionů lidí1. Nevidí tvary nebo dokonce zdroje světla. U většiny těchto lidí je jejich slepota důsledkem napravitelných problémů, jako je šedý zákal – jednoduše nemají přístup k odpovídající zdravotní péči. Zbývající miliony lidí jsou však slepí v důsledku onemocnění, která v současné době nemají účinnou léčbu.
„Slepota je jedním z nejzávažnějších stavů, které mohou člověku změnit život,“ říká William Hauswirth, oftalmolog z Floridské univerzity v Gainesville. Kromě obtíží, které způsobuje při pohybu a hledání zaměstnání, je zrakové postižení spojeno s řadou dalších zdravotních problémů, včetně nespavosti, úzkosti a deprese, a dokonce i s rizikem sebevraždy. „Obnovení užitečného vidění by znamenalo téměř nepředstavitelné zlepšení kvality života,“ říká Hauswirth.
V zemích s vysokými příjmy, kde se běžně řeší příčiny zrakového postižení, kterým lze předcházet, je hlavní příčinou slepoty degenerace sítnice. Tato tkáň, která se nachází v zadní části oka, obsahuje specializované buňky reagující na světlo a zpracovávající zrakové signály, a je tedy pro vidění klíčová. Fotoreceptorové buňky – neurony obecně známé jako tyčinky a čípky – přeměňují světlo dopadající na sítnici na elektrochemické signály. Tyto signály pak procházejí složitou sítí dalších neuronů, včetně bipolárních buněk, amakrinních buněk a horizontálních buněk, než se dostanou k neuronům známým jako gangliové buňky sítnice. Dlouhé výběžky neboli axony těchto buněk tvoří zrakový nerv, po kterém jsou signály ze sítnice přenášeny do zrakové kůry mozku, kde jsou interpretovány jako obrazy.
Poruchy sítnice obvykle zahrnují ztrátu fotoreceptorů, což snižuje citlivost oka na světlo. U některých poruch sítnice, včetně věkem podmíněné makulární degenerace (AMD), je tato ztráta důsledkem selhání epitelových buněk, které tvoří vrstvu v zadní části sítnice známou jako retinální pigmentový epitel (RPE). RPE udržuje buňky fotoreceptorů zdravé tím, že čistí toxické vedlejší produkty vznikající při reakci se světlem a poskytuje jim živiny. Při poruchách sítnice, kdy fotoreceptory zůstávají v dobré kondici, je hlavní příčinou slepoty degenerace gangliových buněk sítnice.
Poslechněte si zvukovou verzi tohoto článku
Váš prohlížeč nepodporuje zvukový prvek.
Různorodost příčin zrakového postižení ztěžuje hledání řešení. Pokroky v několika oblastech však vzbuzují naději, že by se téměř všechny formy poruch sítnice mohly stát léčitelnými.
Jedním z přístupů je rozšíření nebo obejití poškozených očí funkčními protézami. Takové bionické oči mohou v současné době obnovit pouze omezené vidění, ale vědci pokračují v rozšiřování možností těchto zařízení. Další možností je genová terapie. Tento přístup, který je již k dispozici lidem se specifickými genetickými mutacemi, se vědci snaží rozšířit na více lidí a stavů. Někteří vědci se také zabývají léčbou založenou na příbuzné technice známé jako optogenetika, která zahrnuje genetickou změnu buněk s cílem obnovit citlivost sítnice na světlo. Tato práce je v počáteční fázi, ale vědci doufají, že tento přístup bude nakonec schopen pomoci širokému spektru lidí, protože je nezávislý na příčinách degenerace sítnice. A snahy o nahrazení ztracených nebo poškozených buněk sítnice, ať už in situ nebo pomocí transplantace buněk, naznačují, že i pozdní stádia poruch sítnice by nakonec mohla být léčitelná.
Velká část tohoto výzkumu je teprve v plenkách. Hauswirth je však optimistický, pokud jde o pokrok, kterého již bylo dosaženo. Říká, že před deseti lety musel pacientům často říkat, že pro ně nemůže nic udělat. „U mnoha těchto nemocí se to zcela změnilo.“
Bionické oči
Téměř před 30 lety začal Mark Humayun, biomedicínský inženýr z Jihokalifornské univerzity v Los Angeles, elektricky stimulovat sítnice lidí se slepotou. Ve spolupráci s kolegy ze společnosti Second Sight Medical Products, která se zabývá zdravotnickými technologiemi v kalifornském Sylmaru, jeho experimenty ukázaly, že taková stimulace může vyvolat zrakové vnímání světelných skvrn zvaných fosfeny. Po deseti letech práce na zvířatech, jejímž cílem bylo stanovit velikost elektrického proudu, který lze bezpečně aplikovat do oka, a vyzbrojen značně rozšířenými znalostmi o počtu a typech buněk, které přetrvávají v degenerující lidské sítnici, byl Humayunův tým připraven začít pracovat s lidmi. V letech 2002 až 2004 vědci implantovali bionické oko každému ze šesti lidí, kteří měli úplnou nebo téměř úplnou slepotu na jedno oko – jednalo se o první pokus svého druhu. Příjemci zařízení, známého jako Argus I, uváděli, že jsou schopni vnímat fosfeny, směrový pohyb a dokonce i tvary2. Přibližně 300 lidí nyní poznává svět prostřednictvím nástupce tohoto zařízení, přístroje Argus II, který byl v roce 2011 schválen regulačními orgány v Evropě pro použití u lidí s retinitis pigmentosa – skupinou vzácných genetických poruch, které způsobují degeneraci fotoreceptorů. Americký Úřad pro kontrolu potravin a léčiv (FDA) jej následoval o dva roky později.

Implantát Argus II se skládá z pole elektrod, které je připevněno k povrchu sítnice.Kredit: Ringo Chiu/ZUMA /Alamy
Chce-li být pacient vybaven implantátem Argus II, podstoupí operaci, při níž je na povrch sítnice připevněn čip obsahující soustavu elektrod. Pro „vidění“ pomocí zařízení přenáší miniaturní videokamera umístěná na brýlích signály do zpracovatelské jednotky, kterou nosí příjemce. Procesor převádí signály na pokyny, které jsou bezdrátově přenášeny do implantovaného zařízení. Elektrody pak stimulují gangliové buňky sítnice v přední části sítnice. Používání protézy je proces učení. Příjemci musí naučit svůj mozek interpretovat nový typ přijímaných informací. A protože videokamera nesleduje pohyb oka, musí se také naučit pohybovat hlavou, aby nasměrovali svůj pohled.
Zařízení poskytuje pouze omezené vidění. Uživatelé mohou detekovat zdroje světla a objekty s vysoce kontrastními okraji, jako jsou dveře nebo okna, a někteří mohou rozluštit velká písmena. Tato omezení vznikají částečně proto, že 60 elektrod zařízení poskytuje velmi nízké rozlišení ve srovnání s miliony fotoreceptorů ve zdravém oku. Ale i toto minimální vylepšení může lidem výrazně zlepšit život.
Zatímco Argus II je epiretinální implantát – což znamená, že leží na povrchu sítnice – ostatní vyvíjená zařízení jsou navržena tak, aby se umístila pod sítnici. Tyto subretinální implantáty mohou stimulovat buňky, které jsou blíže těm, které normálně přivádějí signály do sítnice – fotoreceptorové buňky. Vědci doufají, že stimulací buněk, které se nacházejí výše ve zrakové dráze, se podaří zachovat větší část zpracování signálů, které provádí zdravá sítnice.
Biotechnologická společnost Retina Implant se sídlem v německém Reutlingenu sestrojila subretinální implantát obsahující fotodiody (polovodičová zařízení, která přeměňují světlo na elektrický proud), které přímo snímají světlo vstupující do oka. To eliminuje potřebu externí videokamery a umožňuje uživatelům přirozeně nasměrovat pohled. Napájení zajišťuje ruční jednotka prostřednictvím cívky, která se implantuje pod kůži nad uchem. Alpha AMS, současná verze systému, získala v Evropě regulační schválení pro použití u lidí s retinitis pigmentosa.
Pixium Vision v Paříži testuje fotovoltaický subretinální implantát nazvaný Prima. Systém promítá signály z videokamery umístěné na brýlích do oka pomocí blízkého infračerveného světla, jehož vlnová délka optimálně pohání fotodiody v zařízení, aby stimulovaly buňky sítnice. Promítání obrazu tímto způsobem dává uživatelům určitou kontrolu nad směrem pohledu, protože mohou zkoumat scénu pouze pohybem očí. Napájení zajišťuje také blízké infračervené světlo, díky čemuž je implantát bezdrátový a operace při jeho montáži méně komplikovaná. „Pacienti se učí, jak rychleji získat zpět zrak, a rozlišení se zdá být lepší,“ říká José-Alain Sahel, oftalmolog z University of Pittsburgh v Pensylvánii, který provádí bezpečnostní testy zařízení u deseti lidí s AMD. „Je to teprve začátek, ale je to velmi slibné.“
Všechna tato zařízení fungují pouze tehdy, když v sítnici zůstávají funkční buňky. U běžných očních onemocnění, která postihují hlavně fotoreceptorové buňky, včetně retinitis pigmentosa a AMD, obvykle nějaké buňky ke stimulaci zbývají. Když však odumře příliš mnoho gangliových buněk sítnice, k čemuž dochází u pokročilé diabetické retinopatie a glaukomu, nemohou tyto implantáty pomoci. Pro lidi bez zbývající funkce sítnice, ať už v důsledku nemoci nebo úrazu, by mohl být vhodnější alternativní bionický přístup.
Humayun a jeho kolegové pracují na systému, který obchází oko a vysílá signály přímo do mozku. Tato myšlenka není nová: v 70. letech 20. století americký biomedicínský inženýr William Dobelle ukázal, že přímá stimulace zrakové kůry vyvolává vnímání fosfenů3. Technologie bionických očí se však dostává do popředí až nyní. Společnost Second Sight vyvinula systém Orion, který je podle Humayuna „v podstatě modifikovaný Argus II“. Podobně jako originál využívá videokameru a signálový procesor, které bezdrátově komunikují s implantátem, ale čip je umístěn na povrchu zrakové kůry, nikoli na sítnici. Zařízení se testuje na pěti lidech s omezeným nebo žádným vnímáním světla v důsledku úrazu oka nebo poškození sítnice či zrakového nervu. „Výsledky jsou zatím dobré,“ říká. „Zatím nás nic nepřekvapilo.“
Vzhledem k tomu, že některé technologie jsou již vyzkoušené a otestované na lidech, je Humayun optimistický a věří, že by systém mohl během několika let získat regulační schválení. „Je zřejmé, že operace mozku má jinou míru rizika, ale postup je poměrně jednoduchý a Orion by mohl pomoci mnohem více pacientům,“ říká. O stimulaci mozku k zajištění užitečného vidění se toho však ví mnohem méně. „Víme toho hodně o sítnici, ale velmi málo o mozkové kůře,“ říká Botond Roska, neurobiolog z Institutu molekulární a klinické oftalmologie ve švýcarské Basileji. „Ale nikdy nebudeme vědět dost, pokud se o to nepokusíme.“
Genová terapie
Oko je ideálním cílem pro genovou terapii. Jelikož je relativně samostatné, viry, které se používají k přenosu genů do buněk sítnice, by neměly být schopny cestovat do jiných částí těla. A protože oko je imunoprivilegovaným místem, je méně pravděpodobné, že by zde imunitní systém proti takovému viru vytvořil obranu.
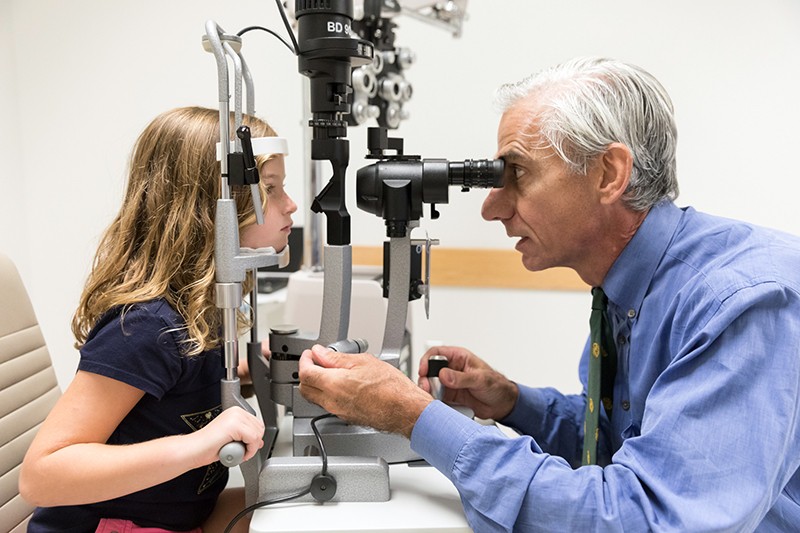
Oftalmolog Albert Maguire prohlíží oči dívky s Leberovou vrozenou amaurózou, které byl zrak navrácen díky genové terapii voretigen neparvovec (Luxturna).Kredit: Dětská nemocnice ve Filadelfii
V první ukázce potenciálu genové terapie při řešení slepoty použily tři týmy vědců tuto techniku k úspěšné léčbě lidí s Leberovou vrozenou amaurózou (LCA). Toto dědičné onemocnění vede k těžkému poškození zraku a začíná v prvních letech života, často se projevuje noční slepotou a poté přechází v rozsáhlou ztrátu zraku, která začíná na periferii zorného pole. Postihuje přibližně 1 ze 40 000 dětí.
Výzkumníci se zaměřili na specifickou formu onemocnění známou jako LCA 2. Ta je způsobena mutacemi v genu RPE65, který je exprimován RPE. Zmutovaný gen nepříznivě ovlivňuje funkci RPE, což následně poškozuje fotoreceptorové buňky. V roce 2008 každý ze tří týmů, včetně týmu vedeného Hauswirthem, prokázal v raných fázích klinických studií, že dodání zdravé kopie RPE65 do sítnice je bezpečné a vede k omezenému zlepšení zraku4,5,6 . Klinická studie fáze III vedená Albertem Maguirem, oftalmologem z Pensylvánské univerzity ve Filadelfii, v srpnu 2017 ukázala, že lidé s LCA 2, kteří podstoupili léčbu, byli schopni lépe se orientovat na překážkových drahách při různých úrovních osvětlení než ti, kteří léčbu nepodstoupili7. V prosinci 2017 schválil úřad FDA léčbu voretigenem neparvovec (Luxturna), čímž se stala první genovou terapií jakéhokoli onemocnění, která dostala zelenou pro klinické použití.
Léčit LCA 2 tímto způsobem je možné, protože příslušné genetické mutace vykazují recesivní vzorec dědičnosti. To znamená, že obě kopie genu RPE65 musí nést příslušné mutace, aby porucha vznikla. Dodání jediné, nemutované verze proto problém řeší. Stavy, které jsou způsobeny dominantně dědičnými mutacemi, však vyžadují pouze jednu mutovanou kopii genu, aby se projevily. U většiny z nich nepomůže pouhé přidání normální kopie genu; namísto toho musí být mutovaný gen inaktivován. Jednou z možností je umlčet ho přidáním specifických molekul RNA, které zachytí instrukce mutovaného genu pro tvorbu vadného proteinu, a poté dodat normální kopii genu, která převezme jeho povinnosti – tento přístup se označuje jako potlačení a nahrazení. Další možností je oprava mutace pomocí techniky úpravy genů CRISPR-Cas9. Výzkumníci z Univerzity v Modeně a Reggio Emilia v italské Modeně demonstrovali tento přístup na myším modelu pigmentové retinitidy8 v roce 2016. V následujícím roce jej tým ve Spojených státech použil k opravě mutace, která způsobuje určitý typ glaukomu, a to jak u myší, tak u kultivovaných lidských buněk9.
Důležitou hnací silou pokroku genové terapie bylo použití adenoasociovaného viru (AAV) k doručení náhradních genů do buněk. Ukázalo se, že AAV jsou bezpečné, částečně proto, že nemají tendenci se integrovat do genomu hostitelské buňky, což minimalizuje riziko, že se buňky stanou rakovinnými. A jejich malá velikost jim umožňuje široce se šířit v oku a infikovat tak velké množství buněk. Schopnost AAV přenášet geny má však své limity: některé geny jsou prostě příliš velké na to, aby je AAV přenášely, včetně genu ABCA4, jehož mutace mohou vést ke Stargardtově chorobě, dědičné formě makulární degenerace. V současné době se hledají dvě řešení. První z nich využívá k přenosu náhradních genů virus s větší nosností, například lentivirus. Bezpečnost a účinnost tohoto přístupu není známa, ale probíhají klinické zkoušky. Druhou strategií je rozdělit náhradní gen na dvě poloviny a každou z nich přenést do buňky zvlášť spolu s prostředkem pro jejich rekombinaci. „To nyní funguje přinejmenším na jednom zvířecím modelu,“ říká Hauswirth.
Bez ohledu na přístup má genová terapie značné omezení. Na vzniku slepoty se podílí více než 250 genů, a protože každý z nich může být postižen mnoha typy mutací, je počet potenciálních terapeutických cílů obrovský. Například více než 100 mutací v genu RHO vede k retinitis pigmentosa, nejčastější dominantně dědičné poruše sítnice. Vývoj genové terapie pro každou jednotlivou mutaci není praktický, říká Hauswirth.
Výzkumníci pracují na možném řešení, které představuje obrat v přístupu „potlačení a nahrazení“. Místo toho, aby se zaměřili na kopie RHO obsahující konkrétní mutaci, používají umlčovací RNA, která potlačí veškerou expresi genu, ať už je RHO mutovaný, nebo ne, a zároveň dodá náhradní kopii, která je vůči umlčovací RNA imunní. Tým vedený Jane Farrarovou, genetičkou z Trinity College v Dublinu, prokázal v roce 2011 nadějnost této strategie na myším modelu dominantní retinitis pigmentosa10. V roce 2018 Hauswirth a jeho kolegové testovali tento přístup u psů s pigmentovou retinitidou11. Ukázali, že degeneraci fotoreceptorových buněk v léčených oblastech sítnice lze zastavit – zlepšení přetrvávalo nejméně osm měsíců. Tato strategie řeší všechny mutace, které mohou způsobit dominantně dědičnou retinitis pigmentosa, jedinou léčbou, a proto rozšiřuje genovou terapii z recesivních na dominantně dědičné stavy „poměrně jednoduchým způsobem“, říká Hauswirth. Plánuje studovat, jak dobře se psi, kteří léčbu podstoupili, dokáží orientovat v bludišti, a shromažďuje údaje o bezpečnosti potřebné k zahájení klinické studie.
Optogenetika
Genová terapie funguje pouze u lidí, jejichž slepota je způsobena genetickou mutací. Není také vhodná pro řešení konečného stádia onemocnění sítnice, ve kterém zbývá nedostatečný počet buněk k opravě. Příbuzný přístup založený na technice zvané optogenetika však není závislý na poruše a mohl by vést k léčbě různých stadií degenerace. Při optogenetice jsou geny, které umožňují buňkám produkovat proteiny citlivé na světlo, známé jako opsiny, dodávány pomocí viru. Zavedením opsinů lze obnovit určitou citlivost poškozených fotoreceptorů na světlo nebo dokonce zvýšit citlivost jiných buněk sítnice, včetně bipolárních buněk nebo gangliových buněk sítnice, na světlo.

Optogenetika byla použita k obnovení citlivosti na světlo u čípkových buněk (zelená) u myšího modelu retinitis pigmentosa; úspěšnost techniky byla hodnocena měřením aktivity gangliové buňky sítnice (purpurová), která je stimulována čípky v reakci na světlo.Kredit: IOB.ch
Problematické však je, že zatímco fotoreceptorové buňky v oku se dokážou vyrovnat se širokým rozsahem intenzity světla – pracují dobře jak za jasného slunečního světla, tak za soumraku – opsiny mají omezený rozsah a často fungují lépe při vysoké intenzitě světla. Možným řešením je použití zařízení, které funguje podobně jako bionický oční systém Prima společnosti Pixium Vision, v němž jsou příjemci vybaveni brýlemi s videokamerou, která snímá pohled uživatele, a projektorem, který míří do jeho oka. Stejně jako u systému Prima je výhodou, že charakter světla, které vstupuje do oka, lze přizpůsobit úpravě sítnice; v tomto případě se však volí intenzita a vlnová délka, které nejlépe řídí nově zavedené opsiny, a nikoli implantované fotodiody.
GenSight Biologics, biotechnologická společnost v Paříži, která mezi své zakladatele počítá Sahela a Rosku, již takový systém testuje. Jejím cílem je dodávat opsin do gangliových buněk sítnice, ale je tu potenciální háček: gangliové buňky sítnice jsou přirozeně citlivé na světlo. Vyjadřují melanopsin, protein, který se podílí na zornicovém světelném reflexu, při němž se oční zornice stahuje v reakci na jasné světlo. Aby k tomu nedocházelo, používají výzkumníci z GenSight opsin, který reaguje na červené vlnové délky světla, protože melanopsin reaguje přednostně na světlo na modrém konci spektra. Společnost zahájila v říjnu 2018 ranou fázi klinické studie u lidí s pokročilou retinitis pigmentosa, kterým zbývá minimum zraku. Studie se zúčastní kohorty ze Spojeného království, Francie a Spojených států a první výsledky se očekávají do konce roku 2020.
„Jedná se o jednoduchý přístup a uvidíme, co se tím získá,“ říká Roska. „Pak můžeme přejít k dalším a sofistikovanějším přístupům.“ Jedním z přetrvávajících problémů je, že mnoho poruch, které by mohly optogenetické techniky léčit, zahrnuje degeneraci specifických částí sítnice, přičemž užitečné vidění zůstává zachováno v jiných oblastech. Světlo, které pohání opsiny, je viditelné a mohlo by narušovat zbývající přirozené vidění. V budoucnu by opsiny reagující na blízké infračervené světlo mohly umožnit, aby optogenetická léčba fungovala společně se zbytky přirozeného zraku.
Buněčná regenerace
Léčba kmenovými buňkami by mohla potenciálně vyléčit slepotu i v pozdních stadiích onemocnění. Protože kmenové buňky lze přimět k tomu, aby se staly jakýmkoli typem buněk, mohly by být použity k vypěstování čerstvých buněk sítnice pro transplantaci do oka, aby nahradily ty, které byly ztraceny. Studie na zvířatech však ukázaly, že jen malá část transplantovaných neuronů se dokáže správně začlenit do složitého nervového obvodu sítnice. To představuje značnou překážku pro léčbu kmenovými buňkami, jejímž cílem je nahradit neurony sítnice.

Složitá buněčná struktura sítnice zahrnuje vrstvy fotoreceptorů (zelená) a cév a nervů (purpurová).Kredit: Louise Hughes/SPL
Buňky tvořící pigmentový epitel sítnice se naopak nacházejí mimo obvody sítnice. Terapie založená na kmenových buňkách je proto nejslibnější u onemocnění, jako je AMD a retinitis pigmentosa, která způsobují degeneraci buněk RPE. „Fotoreceptory musí být napojeny na obvody, ale pigmentový epitel sítnice ne,“ říká Roska. „To je oblast, kde jsou lidé nejblíže pokroku.“ Zpočátku se vědci pokoušeli vstřikovat do sítnice RPE buňky odvozené z kmenových buněk v suspenzi, ale příliš málo jich zůstalo tam, kde bylo potřeba. Několik týmů se nyní domnívá, že lepším přístupem je transplantace buněk RPE do oka ve formě předem připraveného plátu, který je pak udržován na místě biokompatibilním lešením. „Přístup založený na lešení je ve srovnání se suspenzí pro buňky RPE obrovským zlepšením,“ říká Sahel.
V březnu 2018 oznámil London Project to Cure Blindness – spolupráce mezi University College London a Moorfields Eye Hospital v Londýně – výsledky studie fáze I, v níž byl list buněk RPE implantován do sítnice dvou lidí s vlhkým AMD (vzácná, závažná forma AMD zahrnující abnormální růst a únik krevních cév). Oba příjemci snášeli zákrok dobře a byli schopni přečíst na čtecí tabulce o 21-29 písmen více než před léčbou12. Následující měsíc tým vedený Humayunem oznámil podobné výsledky fáze I u pěti osob se suchou formou AMD, což je běžnější forma tohoto onemocnění13. Tyto první výsledky jsou slibné. „Vyvolalo to velké nadšení,“ říká Humayun. Výsledky však musí potvrdit studie fáze III na větším počtu účastníků a Humayun varuje, že od použití této léčby na klinice může uplynout ještě mnoho let, protože žádná terapie kmenovými buňkami pro onemocnění sítnice dosud neprošla schvalovacím procesem.
Související přístup, který je stále v počátečních fázích základního výzkumu, by mohl naplnit naději na nahrazení ztracených neuronů a otevřít tak dveře k léčbě nejrůznějších očních onemocnění. U lidí se zralé neurony nedělí, a proto se nemohou regenerovat, což obnovu zraku obzvlášť ztěžuje. To však neplatí pro všechny živočichy. Plazi a některé ryby mohou regenerovat neurony sítnice a určitou regenerační schopnost vykazují i ptáci. Thomas Reh, neurolog z Washingtonské univerzity v Seattlu, se snaží tuto schopnost odhalit i u lidí. Místo transplantace buněk vypěstovaných v laboratoři se však Reh snaží přimět buňky, které již v sítnici jsou, aby se diferencovaly v nové neurony.
V roce 2001 Reh vyslovil domněnku, že zdrojem nových neuronů, které byly pozorovány u ryb a ptáků, jsou Müllerovy glie – buňky, které zajišťují strukturu sítnice a podporují její funkci14. Se svým týmem se pak pustil do zjišťování, zda lze Müllerovy glie využít k tvorbě čerstvých neuronů u myší. V roce 2015 upravili myši tak, aby vytvářely Ascl1, protein, který je důležitý pro tvorbu neuronů u ryb, a poté zvířatům poškodili sítnici15. Doufali, že Ascl1 vyprovokuje Müllerovy glie k přeměně na neurony.
Pokus nevedl k produkci nových neuronů u dospělých myší, ale uspěl u mladých myší. Nikolas Jorstad, biochemik a doktorand v Rehsově týmu, navrhl, že chemické modifikace chromatinu (komplexu DNA, RNA a proteinů) v buněčném jádře během vývoje mohou v dospělých buňkách blokovat přístup ke genům, které umožňují transformaci Müllerových glií na neurony. V srpnu 2017 Rehův tým ukázal, že zavedením enzymu, který tyto modifikace zvrátí, může přimět Müllerovy glie k diferenciaci16. „Poprvé jsme tak mohli regenerovat neurony u dospělé myši,“ říká Reh. „Po všech těch letech jsem byl docela nadšený.“ I když to nebyly pravé fotoreceptorové buňky a vypadaly spíše jako bipolární buňky, neurony se napojily na stávající obvody a byly citlivé na světlo. „Překvapilo mě, že se propojují tak dobře, jak se propojují,“ říká Reh.
Ačkoli tato práce ještě zdaleka není připravena k léčbě poruch sítnice u lidí, má obrovský potenciál. Dalším krokem bude zopakování studií na zvířatech s očima, které jsou více podobné těm lidským. Rehův tým již pracuje s buněčnými kulturami sítnice primátů, kteří nejsou lidmi. Vědci také musí přijít na to, jak řídit proces diferenciace tak, aby produkoval specifické typy buněk, jako jsou tyčinky a čípky. „Teď už máme našlápnuto k výrobě neuronů, čípky by byly skvělé,“ říká Reh.
Pokud bude tento přístup úspěšný, mohl by být široce použitelný. „Nakonec se tímto způsobem budou léčit všechny tyto oční choroby,“ předpovídá Reh. „Dává to prostě smysl. Nemusíte se starat o správné provedení transplantace. Vaše buňky jsou přesně tam, kde je potřebujete.“
Humayun je touto prací také povzbuzen. „Fandím každému, kdo má nový dobrý nápad,“ říká. „Je to velmi brzy, je to vysoce rizikové, ale nikdy neříkej nikdy. To jsem se naučil.“