En todo el mundo, 36 millones de personas tienen pérdida total de visión1. No pueden ver las formas ni siquiera las fuentes de luz. Para la mayoría de estas personas, su ceguera se debe a problemas rectificables, como las cataratas; simplemente carecen de acceso a una atención sanitaria adecuada. Los millones restantes, sin embargo, son ciegos como resultado de condiciones que actualmente no tienen un tratamiento efectivo.
«La ceguera es una de las condiciones que más cambian la vida de una persona», dice William Hauswirth, oftalmólogo de la Universidad de Florida en Gainesville. Además de las dificultades que provoca para la movilidad y la búsqueda de empleo, la discapacidad visual se asocia a una serie de otros problemas de salud, como el insomnio, la ansiedad y la depresión, e incluso el riesgo de suicidio. «Restablecer la visión útil supondría una mejora casi inimaginable de la calidad de vida», afirma Hauswirth.
En los países de ingresos altos, donde las causas prevenibles de la discapacidad visual se abordan de forma rutinaria, la principal causa de ceguera es la degeneración de la retina. Este tejido, situado en la parte posterior del ojo, contiene células especializadas que reaccionan a la luz y procesan las señales visuales, por lo que es crucial para la visión. Las células fotorreceptoras – neuronas comúnmente conocidas como bastones y conos – convierten la luz que incide en la retina en señales electroquímicas. Estas señales se filtran a continuación a través de una compleja red de otras neuronas, como las células bipolares, las células amacrinas y las células horizontales, antes de llegar a las neuronas conocidas como células ganglionares de la retina. Las largas proyecciones, o axones, de esas células forman el nervio óptico, a lo largo del cual las señales de la retina son llevadas a la corteza visual del cerebro, donde se interpretan como imágenes.
Los trastornos de la retina suelen implicar la pérdida de células fotorreceptoras, lo que agota la sensibilidad del ojo a la luz. En algunos trastornos de la retina, como la degeneración macular asociada a la edad (DMAE), esta pérdida se produce por el fallo de las células epiteliales que forman una capa en la parte posterior de la retina conocida como epitelio pigmentario de la retina (EPR). El EPR mantiene sanas a las células fotorreceptoras limpiando los subproductos tóxicos producidos durante la reacción con la luz, así como aportando nutrientes. En los trastornos de la retina en los que los fotorreceptores se mantienen en buen estado, la principal causa de ceguera es la degeneración de las células ganglionares de la retina.
Escucha una versión de audio de este artículo
Tu navegador no admite el elemento de audio.
La variedad en las causas de la discapacidad visual hace más difícil encontrar soluciones. Pero los avances en varias áreas hacen esperar que casi todas las formas de trastorno de la retina puedan llegar a ser tratables.
Una aproximación es aumentar o puentear los ojos dañados con prótesis funcionales. En la actualidad, estos ojos biónicos sólo pueden restablecer una visión limitada, pero los investigadores siguen ampliando las capacidades de estos dispositivos. Otra opción es la terapia génica. Ya disponible para personas con mutaciones genéticas específicas, los investigadores quieren ampliar este enfoque a más personas y afecciones. Algunos científicos también están buscando tratamientos basados en una técnica relacionada conocida como optogenética, que consiste en alterar genéticamente las células para restaurar la sensibilidad a la luz en la retina. Este trabajo se encuentra en una fase inicial, pero los investigadores esperan que este método pueda ayudar a un gran número de personas, ya que es independiente de las causas de la degeneración de la retina. Y los esfuerzos por sustituir las células perdidas o dañadas de la retina, ya sea in situ o mediante trasplantes celulares, insinúan que incluso los trastornos de la retina en fase avanzada podrían llegar a ser tratables.
Mucha de esta investigación está en sus inicios. Sin embargo, Hauswirth es optimista respecto a los progresos que ya se han realizado. Hace diez años, dice, a menudo tenía que decir a los pacientes que no podía hacer nada por ellos. «Para muchas de estas enfermedades, eso ha cambiado totalmente».
Ojos biónicos
Hace casi 30 años, Mark Humayun, ingeniero biomédico de la Universidad del Sur de California en Los Ángeles, empezó a estimular eléctricamente las retinas de personas con ceguera. En colaboración con sus colegas de Second Sight Medical Products, una empresa de tecnología médica de Sylmar (California), sus experimentos demostraron que dicha estimulación podía inducir la percepción visual de unos puntos de luz llamados fosfenos. Tras una década de trabajo en animales para establecer la cantidad de corriente eléctrica que podía aplicarse con seguridad al ojo, y armados con un conocimiento mucho mayor sobre el número y los tipos de células que persisten en las retinas humanas degeneradas, el equipo de Humayun estaba listo para empezar a trabajar con personas. Entre 2002 y 2004, los investigadores implantaron un ojo biónico en cada una de las seis personas que padecían ceguera total o casi total en un ojo, lo que supuso el primer ensayo de este tipo. Los receptores del dispositivo, conocido como Argus I, declararon ser capaces de percibir fosfenos, movimientos direccionales e incluso formas2. Alrededor de 300 personas experimentan ahora el mundo a través del sucesor de ese dispositivo, el Argus II, que fue aprobado por los reguladores en Europa en 2011 para su uso en personas con retinosis pigmentaria, un grupo de trastornos genéticos raros que causan la degeneración de las células fotorreceptoras. La Administración de Alimentos y Medicamentos de Estados Unidos (FDA) hizo lo propio dos años después.

El implante Argus II consta de una matriz de electrodos que se fija a la superficie de la retina.Crédito: Ringo Chiu/ZUMA /Alamy
Para que se les coloque un Argus II, los pacientes se someten a una intervención quirúrgica en la que se fija un chip que contiene un conjunto de electrodos en la superficie de la retina. Para «ver» con el dispositivo, una cámara de vídeo en miniatura montada en unas gafas transmite señales a una unidad de procesamiento que lleva el receptor. El procesador convierte las señales en instrucciones que se transmiten de forma inalámbrica al dispositivo implantado. Los electrodos estimulan entonces las células ganglionares de la parte delantera de la retina. El uso de la prótesis es un proceso de aprendizaje. Los receptores deben entrenar su cerebro para interpretar el nuevo tipo de información que reciben. Y como la cámara de vídeo no sigue el movimiento del ojo, también deben aprender a mover la cabeza para dirigir la mirada.
El dispositivo sólo proporciona una visión limitada. Los usuarios pueden detectar fuentes de luz y objetos con bordes de alto contraste, como puertas o ventanas, y algunos pueden descifrar letras grandes. Estas limitaciones se deben en parte a que los 60 electrodos del dispositivo proporcionan una resolución muy baja en comparación con los millones de células fotorreceptoras de un ojo sano. Pero incluso esta mínima mejora puede mejorar considerablemente la vida de las personas.
Mientras que el Argus II es un implante epirretiniano -lo que significa que se sitúa en la superficie de la retina-, otros dispositivos en desarrollo están diseñados para colocarse debajo de la retina. Estos implantes subretinianos pueden estimular células que están más cerca de las que normalmente introducen señales en la retina: las células fotorreceptoras. Al estimular células situadas más arriba en la vía visual, los investigadores esperan conservar una mayor parte del procesamiento de señales que realiza una retina sana.
Retina Implant, una empresa de biotecnología con sede en Reutlingen (Alemania), ha construido un implante subretiniano compuesto por fotodiodos (dispositivos semiconductores que convierten la luz en corriente eléctrica) que detectan directamente la luz que entra en el ojo. Esto elimina la necesidad de una cámara de vídeo externa, lo que permite a los usuarios dirigir su mirada de forma natural. La energía la suministra una unidad manual, a través de una bobina que se implanta bajo la piel, encima de la oreja. Alpha AMS, la versión actual del sistema, ha recibido la aprobación reglamentaria en Europa para su uso en personas con retinosis pigmentaria.
Pixium Vision, en París, está probando un implante subretiniano fotovoltaico llamado Prima. El sistema proyecta en el ojo señales procedentes de una cámara de vídeo montada en unas gafas utilizando luz infrarroja cercana, cuya longitud de onda impulsa de forma óptima los fotodiodos del dispositivo para estimular las células de la retina. La proyección de imágenes de este modo permite a los usuarios controlar la dirección de su mirada, ya que pueden explorar la escena moviendo sólo los ojos. La luz infrarroja cercana también proporciona energía, lo que hace que el implante sea inalámbrico y la cirugía para colocarlo menos complicada. «Los pacientes están aprendiendo a recuperar la visión más rápidamente, y la resolución parece mejor», dice José-Alain Sahel, oftalmólogo de la Universidad de Pittsburgh (Pensilvania), que está realizando ensayos de seguridad del dispositivo en diez personas con DMAE. «Es pronto, pero es muy prometedor».
Todos estos dispositivos funcionan sólo cuando quedan células funcionales en la retina. En las enfermedades oculares comunes que afectan principalmente a las células fotorreceptoras, como la retinosis pigmentaria y la DMAE, suelen quedar algunas células para estimular. Pero cuando mueren demasiadas células ganglionares de la retina, como ocurre en la retinopatía diabética avanzada y el glaucoma, estos implantes no pueden ayudar. Para las personas a las que no les queda ninguna función retiniana, ya sea por enfermedad o por lesión, podría ser más pertinente un enfoque biónico alternativo.
Humayun y sus colegas están trabajando en un sistema que evita el ojo enviando las señales directamente al cerebro. La idea no es nueva: en la década de 1970, el ingeniero biomédico estadounidense William Dobelle demostró que la estimulación directa de la corteza visual provocaba la percepción de fosfenos3. Pero la tecnología de los ojos biónicos se está poniendo al día. Second Sight ha desarrollado Orion, un sistema que es, según Humayun, «básicamente un Argus II modificado». Al igual que el original, utiliza una cámara de vídeo y un procesador de señales que se comunican de forma inalámbrica con un implante, pero el chip se coloca en la superficie de la corteza visual en lugar de en la retina. El dispositivo se está probando en cinco personas con percepción de la luz limitada o nula debido a una lesión ocular o a daños en la retina o el nervio óptico. «Hasta ahora, los resultados son buenos», dice. «Todavía no nos sorprende nada».
Dado que parte de la tecnología ya está probada en personas, Humayun es optimista y cree que el sistema podría recibir la aprobación reglamentaria dentro de unos años. «Obviamente, la cirugía cerebral tiene un nivel de riesgo diferente, pero el procedimiento es bastante sencillo, y el Orion podría ayudar a muchos más pacientes», afirma. Sin embargo, se sabe mucho menos sobre la estimulación del cerebro para proporcionar una visión útil. «Sabemos mucho sobre la retina, pero muy poco sobre el córtex», afirma Botond Roska, neurobiólogo del Instituto de Oftalmología Molecular y Clínica de Basilea (Suiza). «Pero nunca sabremos lo suficiente si no lo intentamos», afirma.
Terapia génica
El ojo es un objetivo ideal para la terapia génica. Al ser relativamente autónomo, los virus que se utilizan para llevar los genes a las células de la retina no deberían poder viajar a otras partes del cuerpo. Y como el ojo es un lugar inmunoprivilegiado, es menos probable que el sistema inmunitario monte allí una defensa contra un virus de este tipo.
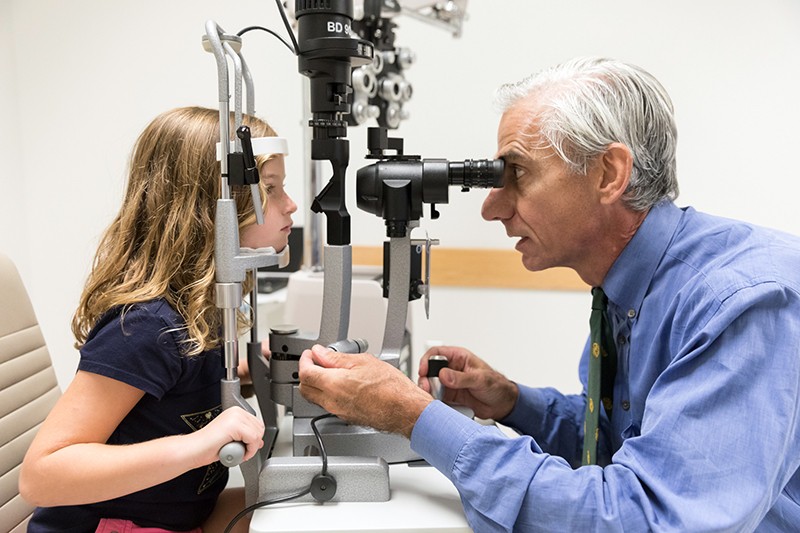
El oftalmólogo Albert Maguire examina los ojos de una niña con amaurosis congénita de Leber a la que se le ha devuelto la vista gracias a la terapia génica voretigene neparvovec (Luxturna).Crédito: Hospital Infantil de Filadelfia
En la primera demostración del potencial de la terapia génica para abordar la ceguera, tres equipos de investigadores han utilizado la técnica para tratar con éxito a personas con amaurosis congénita de Leber (ACL). Esta enfermedad hereditaria provoca una grave deficiencia visual que comienza en los primeros años de vida y suele manifestarse como ceguera nocturna antes de progresar a una pérdida de visión amplia que comienza en la periferia del campo visual. Afecta a aproximadamente 1 de cada 40.000 bebés.
Los investigadores se propusieron abordar una forma específica de la enfermedad conocida como LCA 2. Ésta está causada por mutaciones en el RPE65, un gen que se expresa en el RPE. El gen mutado afecta negativamente a la función del EPR, lo que a su vez daña las células fotorreceptoras. En 2008, los tres equipos, incluido el dirigido por Hauswirth, demostraron en ensayos clínicos de fase inicial que la administración de una copia sana de RPE65 en la retina era segura y producía mejoras limitadas en la visión4,5,6. Un ensayo clínico de fase III dirigido por Albert Maguire, oftalmólogo de la Universidad de Pensilvania en Filadelfia, demostró en agosto de 2017 que las personas con LCA 2 que recibieron el tratamiento eran más capaces de navegar por carreras de obstáculos en varios niveles de iluminación que los que no lo hicieron7. En diciembre de 2017, la FDA aprobó el tratamiento, voretigene neparvovec (Luxturna), convirtiéndolo en la primera terapia génica para cualquier condición en obtener luz verde para su uso clínico.
Es posible tratar la LCA 2 de esta manera porque las mutaciones genéticas involucradas muestran un patrón de herencia recesivo. Esto significa que las dos copias de RPE65 de una persona deben ser portadoras de las mutaciones pertinentes para causar el trastorno. Por lo tanto, el suministro de una única versión no mutada soluciona el problema. Sin embargo, las enfermedades causadas por mutaciones de herencia dominante sólo requieren una copia mutada de un gen para manifestarse. En la mayoría de estos casos, la simple adición de una copia normal del gen no servirá de nada, sino que habrá que inactivar el gen mutado. Una opción es silenciarlo añadiendo moléculas de ARN específicas que intercepten las instrucciones del gen mutado para fabricar la proteína defectuosa y, a continuación, suministrar una copia normal del gen para que asuma sus funciones, un enfoque denominado supresión y sustitución. Otro método consiste en corregir la mutación mediante la técnica de edición de genes CRISPR-Cas9. Investigadores de la Universidad de Módena y Reggio Emilia en Módena (Italia) demostraron este método en un modelo de ratón de retinosis pigmentaria8 en 2016. Al año siguiente, un equipo de Estados Unidos lo utilizó para corregir la mutación que causa un tipo de glaucoma tanto en ratones como en células humanas cultivadas9.
Un importante motor del progreso de la terapia génica ha sido el uso del virus adeno-asociado (AAV) para suministrar genes de sustitución a las células. Los AAV han demostrado ser seguros, en parte, porque tienden a no integrarse en el genoma de su célula huésped, lo que minimiza el riesgo de que las células se vuelvan cancerosas. Además, su pequeño tamaño les permite difundirse ampliamente por el ojo y, por tanto, infectar un gran número de células. Pero la capacidad de los AAV para transportar genes tiene límites: algunos genes son demasiado grandes para los AAV, como el ABCA4, cuyas mutaciones pueden provocar la enfermedad de Stargardt, una forma hereditaria de degeneración macular. Se están buscando dos soluciones. La primera consiste en utilizar un virus con mayor capacidad de transporte, como un lentivirus, para administrar los genes de sustitución. Se desconoce la seguridad y eficacia de este método, pero se están realizando ensayos clínicos. Una segunda estrategia consiste en dividir el gen de sustitución en dos y transportar cada mitad por separado al interior de la célula, junto con un medio para recombinarlas. «Eso está funcionando en al menos un modelo animal ahora mismo», dice Hauswirth.
Independientemente del enfoque, la terapia génica tiene una limitación considerable. Hay más de 250 genes implicados en la ceguera y, dado que cada uno de ellos puede verse afectado por numerosos tipos de mutación, el número de objetivos terapéuticos potenciales es enorme. Por ejemplo, más de 100 mutaciones en el gen RHO dan lugar a la retinosis pigmentaria, el trastorno de la retina de herencia dominante más frecuente. Desarrollar una terapia génica para todas y cada una de las mutaciones no es práctico, dice Hauswirth.
Los investigadores están trabajando en una posible solución que da un giro al enfoque de supresión y sustitución. En lugar de dirigirse a las copias de RHO que contienen una mutación específica, utilizan un ARN silenciador para suprimir toda la expresión del gen, tanto si RHO está mutado como si no, al tiempo que entregan una copia de reemplazo que es inmune al ARN silenciador. Un equipo dirigido por Jane Farrar, genetista del Trinity College de Dublín, mostró la promesa de esta estrategia en 2011 en un modelo de ratón de retinosis pigmentaria dominante10. En 2018, Hauswirth y sus colegas probaron el enfoque en perros con retinosis pigmentaria11. Demostraron que se podía detener la degeneración de las células fotorreceptoras en las zonas de la retina tratadas, una mejora que persistió durante al menos ocho meses. Esta estrategia aborda todas las mutaciones que pueden causar retinosis pigmentaria de herencia dominante en un único tratamiento y, por tanto, amplía la terapia génica de las afecciones recesivas a las de herencia dominante «de una forma bastante sencilla», afirma Hauswirth. Tiene previsto estudiar la capacidad de los perros que han recibido el tratamiento para recorrer un laberinto, y está recopilando los datos de seguridad necesarios para iniciar un ensayo clínico.
Optogenética
La terapia génica sólo funciona en personas cuya ceguera está causada por una mutación genética. Tampoco es adecuada para abordar la enfermedad de la retina en fase terminal, en la que queda un número insuficiente de células para reparar. Pero un enfoque relacionado, basado en una técnica llamada optogenética, es agnóstico respecto a los trastornos y podría dar lugar a tratamientos para diferentes etapas de la degeneración. En la optogenética, los genes que permiten a las células producir proteínas sensibles a la luz, conocidas como opsinas, se introducen mediante un virus. La introducción de opsinas puede restaurar parte de la sensibilidad a la luz de los fotorreceptores dañados, o incluso hacer que otras células de la retina, incluidas las células bipolares o las células ganglionares de la retina, sean sensibles a la luz.

Se ha utilizado la ontogenia para restaurar la sensibilidad a la luz de las células de los conos (verde) en un modelo de ratón de retinosis pigmentaria; el éxito de la técnica se evaluó midiendo la actividad de una célula ganglionar de la retina (magenta), que es estimulada por los conos en respuesta a la luz.Crédito: IOB.ch
Sin embargo, mientras que las células fotorreceptoras del ojo pueden hacer frente a una amplia gama de intensidades de luz -funcionan bien tanto a la luz del sol como en el crepúsculo-, las opsinas tienen un rango limitado y a menudo funcionan mejor a altas intensidades de luz. Una posible solución es utilizar un sistema similar al del ojo biónico Prima de Pixium Vision, en el que los receptores llevan unas gafas que incorporan una cámara de vídeo que capta la vista del usuario y un proyector que apunta a su ojo. Al igual que con Prima, la ventaja es que la naturaleza de la luz que entra en el ojo puede adaptarse a la modificación de la retina; sin embargo, en este caso, la intensidad y la longitud de onda elegidas son las que mejor impulsan las opsinas recién introducidas en lugar de los fotodiodos implantados.
GenSight Biologics, una empresa de biotecnología de París que cuenta con Sahel y Roska entre sus fundadores, ya está probando un sistema de este tipo. Su objetivo es suministrar una opsina a las células ganglionares de la retina, pero existe un posible inconveniente: las células ganglionares de la retina son sensibles a la luz por naturaleza. Expresan melanopsina, una proteína que interviene en el reflejo pupilar de la luz, en el que la pupila del ojo se contrae en respuesta a la luz brillante. Para evitarlo, los investigadores de GenSight utilizan una opsina que responde a las longitudes de onda del rojo, ya que la melanopsina responde preferentemente a la luz del extremo azul del espectro. La compañía comenzó un ensayo clínico en fase inicial en octubre de 2018 en personas con retinosis pigmentaria avanzada a las que les queda una vista mínima. En el ensayo participarán cohortes de Reino Unido, Francia y Estados Unidos, y los resultados iniciales se esperan para finales de 2020.
«Se trata de un enfoque sencillo, y tendremos que ver qué se gana», afirma Roska. «Entonces, podremos pasar a enfoques más y más sofisticados». Un problema que persiste es que muchos de los trastornos que podrían tratar las técnicas optogenéticas implican la degeneración de partes específicas de la retina, conservando la visión útil en otras zonas. La luz que impulsa las opsinas es visible y podría interferir con la visión natural restante. En el futuro, las opsinas que responden a la luz cercana al infrarrojo podrían permitir que los tratamientos optogenéticos funcionaran junto con la visión natural residual.
Regeneración celular
La terapia con células madre podría curar la ceguera incluso en las últimas fases de la enfermedad. Dado que las células madre pueden convertirse en cualquier tipo de célula, podrían utilizarse para cultivar nuevas células de la retina y trasplantarlas al ojo para reemplazar las que se han perdido. Sin embargo, los estudios realizados en animales han demostrado que sólo una pequeña proporción de las neuronas trasplantadas son capaces de integrarse correctamente en el complejo circuito neuronal de la retina. Esto supone un obstáculo considerable para los tratamientos con células madre que pretenden sustituir las neuronas de la retina.

La compleja estructura celular de la retina incluye capas de fotorreceptores (verde) y vasos sanguíneos y nervios (magenta).Crédito: Louise Hughes/SPL
Las células que componen el epitelio pigmentario de la retina, por otra parte, se sitúan fuera de los circuitos de la retina. Por lo tanto, las terapias basadas en células madre son más prometedoras para enfermedades, como la DMAE y la retinosis pigmentaria, que provocan la degeneración de las células del EPR. «Los fotorreceptores tienen que conectarse a los circuitos, pero el epitelio pigmentario de la retina no», dice Roska. «Ahí es donde más se está avanzando». Al principio, los investigadores intentaron inyectar en la retina células del EPR derivadas de células madre en suspensión, pero muy pocas se quedaban donde se necesitaban. Varios equipos piensan ahora que un enfoque mejor es trasplantar las células del EPR en el ojo como una lámina preformada que luego se mantiene en posición mediante un andamio biocompatible. «El enfoque del andamio supone una enorme mejora, en comparación con la suspensión, para las células del EPR», afirma Sahel.
En marzo de 2018, el Proyecto de Londres para Curar la Ceguera -una colaboración entre el University College London y el Moorfields Eye Hospital de Londres- anunció los resultados de un ensayo de fase I en el que se implantó una lámina de células del EPR en las retinas de dos personas con DMAE húmeda (una forma rara y grave de DMAE que implica el crecimiento anormal y la fuga de vasos sanguíneos). Ambos receptores toleraron bien el procedimiento y fueron capaces de leer entre 21 y 29 letras más en una tabla de lectura que antes del tratamiento12. Al mes siguiente, un equipo dirigido por Humayun comunicó resultados similares de fase I en cinco personas con DMAE seca, la forma más común de la enfermedad13. Estos resultados iniciales son muy prometedores. «Esto ha provocado un gran entusiasmo», afirma Humayun. Pero los hallazgos deben ser confirmados por ensayos de fase III en un mayor número de participantes, y Humayun advierte que el tratamiento podría estar a muchos años de distancia de su uso en la clínica, porque ninguna terapia con células madre para un trastorno de la retina ha superado aún el proceso de aprobación.
Un enfoque relacionado, todavía en las primeras etapas de la investigación básica, podría cumplir la esperanza de reemplazar las neuronas perdidas, abriendo la puerta a tratamientos para una amplia variedad de enfermedades oculares. En los seres humanos, las neuronas maduras no se dividen y, por tanto, no pueden regenerarse, lo que dificulta especialmente la restauración de la visión. Pero no ocurre lo mismo en todos los animales. Los reptiles y algunos peces pueden regenerar las neuronas de la retina, y las aves también presentan cierta capacidad de regeneración. Thomas Reh, neurocientífico de la Universidad de Washington, en Seattle, está tratando de desarrollar esta capacidad en los seres humanos. Pero en lugar de trasplantar células cultivadas en el laboratorio, Reh pretende convencer a las células que ya están en la retina para que se diferencien en nuevas neuronas.
En 2001, Reh sugirió que la glía de Müller -células que proporcionan estructura a la retina y apoyan su función- es la fuente de nuevas neuronas que se había observado en peces y aves14. Él y su equipo se propusieron entonces averiguar si la glía de Müller podía utilizarse para generar nuevas neuronas en ratones. En 2015, diseñaron ratones para que produjeran Ascl1, una proteína importante para la producción de neuronas en los peces, y luego dañaron las retinas de los animales15. Su esperanza era que Ascl1 provocara que la glía de Müller se transformara en neuronas.
El experimento no consiguió producir nuevas neuronas en ratones adultos, pero sí en ratones jóvenes. Nikolas Jorstad, bioquímico y estudiante de doctorado del equipo de Rehs, propuso que las modificaciones químicas realizadas en la cromatina (un complejo de ADN, ARN y proteínas) en el núcleo celular durante el desarrollo podrían bloquear el acceso en las células maduras a los genes que permiten a la glía de Müller transformarse en neuronas. En agosto de 2017, el equipo de Reh demostró que, introduciendo una enzima que revierte dichas modificaciones, podían engatusar a la glía de Müller para que se diferenciara16. «Por primera vez, pudimos regenerar neuronas en el ratón adulto», dice Reh. «Después de tantos años, estaba muy emocionado». Aunque no eran verdaderas células fotorreceptoras y parecían más bien células bipolares, las neuronas se conectaban al circuito existente y eran sensibles a la luz. «Me sorprendió que se conectaran tan bien como lo hacen», dice Reh.
Aunque está lejos de estar listo para tratar los trastornos de la retina en las personas, el trabajo tiene un enorme potencial. El siguiente paso será repetir los estudios en animales con ojos más parecidos a los de los humanos. El equipo de Reh ya está trabajando con cultivos de células de la retina de primates no humanos. Los investigadores también tienen que averiguar cómo dirigir el proceso de diferenciación para producir tipos celulares específicos, como bastones y conos. «Ahora que hemos metido el pie en el negocio de la fabricación de neuronas, los conos serían geniales», dice Reh.
Si tiene éxito, el enfoque podría ser ampliamente aplicable. «En última instancia, esta será la forma de tratar todas estas enfermedades oculares», predice Reh. «Simplemente tiene sentido. No hay que preocuparse por hacer bien los trasplantes. Tus células están justo donde las necesitas».
Humayun también está animado por el trabajo. «Aplaudo a cualquiera que tenga una nueva buena idea», dice. «Es muy pronto, es de alto riesgo, pero nunca digas nunca. Eso es lo que he aprendido»