Takeaways
-
Un logiciel d' »apprentissage profond » du laboratoire DeepMind, propriété de Google, a montré de grands progrès dans la résolution de l’un des plus grands défis de la biologie – comprendre le repliement des protéines.
-
Le repliement des protéines est le processus par lequel une protéine prend sa forme à partir d’une chaîne de blocs de construction jusqu’à sa structure tridimensionnelle finale, qui détermine sa fonction.
-
En prédisant mieux comment les protéines prennent leur structure, ou « se plient », les scientifiques peuvent développer plus rapidement des médicaments qui, par exemple, bloquent l’action de protéines virales cruciales.
Résoudre ce que les biologistes appellent « le problème du repliement des protéines » est une affaire importante. Les protéines sont les chevaux de trait des cellules et sont présentes dans tous les organismes vivants. Elles sont constituées de longues chaînes d’acides aminés et sont vitales pour la structure des cellules et la communication entre elles, ainsi que pour la régulation de toute la chimie du corps.
Cette semaine, DeepMind, société d’intelligence artificielle appartenant à Google, a fait la démonstration d’un programme d’apprentissage profond appelé AlphaFold2, que les experts qualifient de percée vers la résolution du grand défi du repliement des protéines.
Les protéines sont de longues chaînes d’acides aminés reliées entre elles comme des perles sur un fil. Mais pour qu’une protéine fasse son travail dans la cellule, elle doit se « plier » – un processus de torsion et de pliage qui transforme la molécule en une structure tridimensionnelle complexe capable d’interagir avec sa cible dans la cellule. Si le repliement est perturbé, la protéine ne prendra pas la forme correcte et ne sera pas en mesure d’accomplir sa tâche dans l’organisme. Cela peut entraîner une maladie – comme c’est le cas dans une maladie courante comme Alzheimer, et dans des maladies rares comme la mucoviscidose.
L’apprentissage profond est une technique informatique qui utilise les informations souvent cachées contenues dans de vastes ensembles de données pour résoudre des questions d’intérêt. Elle a été largement utilisée dans des domaines tels que les jeux, la reconnaissance vocale et de la parole, les voitures autonomes, la science et la médecine.
Je pense que des outils comme AlphaFold2 aideront les scientifiques à concevoir de nouveaux types de protéines, celles qui pourraient, par exemple, aider à décomposer les plastiques et à lutter contre les futures pandémies virales et maladies.
Je suis un chimiste computationnel et l’auteur du livre The State of Science. Mes étudiants et moi étudions la structure et les propriétés des protéines fluorescentes à l’aide de programmes informatiques de repliement des protéines basés sur la physique classique.
Après des décennies d’étude par des milliers de groupes de recherche, ces programmes de prédiction du repliement des protéines sont très bons pour calculer les changements structurels qui se produisent lorsque nous apportons de petites modifications à des molécules connues.
Mais ils n’ont pas réussi de manière adéquate à prédire comment les protéines se replient à partir de zéro. Avant l’arrivée de l’apprentissage profond, le problème du repliement des protéines semblait incroyablement difficile, et il semblait prêt à frustrer les chimistes computationnels pour de nombreuses décennies à venir.
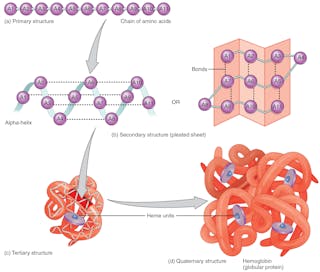
Pliage des protéines
La séquence des acides aminés – qui est codée dans l’ADN – définit la forme 3D de la protéine. Cette forme détermine sa fonction. Si la structure de la protéine change, elle est incapable de remplir sa fonction. Prédire correctement les plis des protéines sur la base de la séquence d’acides aminés pourrait révolutionner la conception des médicaments, et expliquer les causes de maladies nouvelles et anciennes.
Toutes les protéines ayant la même séquence de blocs de construction d’acides aminés se replient dans la même forme tridimensionnelle, qui optimise les interactions entre les acides aminés. Elles y parviennent en quelques millisecondes, bien qu’elles aient à leur disposition un nombre astronomique de configurations possibles – environ 10 à la puissance 300. C’est en raison de ce nombre considérable qu’il est difficile de prédire comment une protéine se replie, même lorsque les scientifiques connaissent la séquence complète des acides aminés qui entrent dans sa composition. Auparavant, il était impossible de prédire la structure d’une protéine à partir de la séquence d’acides aminés. Les structures protéiques étaient déterminées expérimentalement, une entreprise longue et coûteuse.
Une fois que les chercheurs pourront mieux prédire comment les protéines se replient, ils seront en mesure de mieux comprendre le fonctionnement des cellules et comment les protéines mal repliées provoquent des maladies. De meilleurs outils de prédiction des protéines nous aideront également à concevoir des médicaments capables de cibler une région topologique particulière d’une protéine où se produisent des réactions chimiques.

AlphaFold est né de l’apprentissage profond des jeux d’échecs, de Go et de poker
Le succès du programme de prédiction du repliement des protéines de DeepMind, appelé AlphaFold, n’est pas inattendu. D’autres programmes d’apprentissage profond écrits par DeepMind ont démoli les meilleurs joueurs d’échecs, de Go et de poker du monde.
En 2016, Stockfish-8, un moteur d’échecs open-source, était le champion du monde d’échecs par ordinateur. Il évaluait 70 millions de positions échiquéennes par seconde et pouvait s’appuyer sur des siècles de stratégies échiquéennes humaines accumulées et des décennies d’expérience informatique. Il jouait de manière efficace et brutale, battant impitoyablement tous ses adversaires humains sans une once de finesse. Entrez dans l’apprentissage profond.
Le 7 décembre 2017, AlphaZero, le programme d’échecs à apprentissage profond de Google, a battu Stockfish-8. Les moteurs d’échecs ont joué 100 parties, AlphaZero en a gagné 28 et fait match nul 72. Il n’a pas perdu une seule partie. AlphaZero n’a effectué que 80 000 calculs par seconde, contre les 70 millions de calculs de Stockfish-8, et il lui a fallu seulement quatre heures pour apprendre les échecs à partir de zéro en jouant contre lui-même quelques millions de fois et en optimisant ses réseaux neuronaux au fur et à mesure qu’il apprenait de son expérience.
AlphaZero n’a rien appris des humains ou des parties d’échecs jouées par des humains. Il s’est enseigné lui-même et, ce faisant, a dérivé des stratégies jamais vues auparavant. Dans un commentaire du magazine Science, l’ancien champion du monde d’échecs Garry Kasparov a écrit qu’en apprenant à jouer lui-même, AlphaZero a développé des stratégies qui « reflètent la vérité » des échecs plutôt que de refléter « les priorités et les préjugés » des programmeurs. « C’est l’incarnation du cliché ‘travailler plus intelligemment, pas plus dur' ».
CASP – les Jeux olympiques pour les modélisateurs moléculaires
Tous les deux ans, les meilleurs chimistes informaticiens du monde testent les capacités de leurs programmes à prédire le repliement des protéines et participent au concours CASP (Critical Assessment of Structure Prediction).
Dans le cadre de ce concours, les équipes reçoivent la séquence linéaire d’acides aminés d’une centaine de protéines dont la forme 3D est connue mais n’a pas encore été publiée ; elles doivent ensuite calculer comment ces séquences se replieraient. En 2018, AlphaFold, le débutant en apprentissage profond de la compétition, a battu tous les programmes traditionnels – mais de justesse.
Deux ans plus tard, lundi, on a annoncé qu’Alphafold2 avait remporté le concours de 2020 avec une bonne marge. Il a fouetté ses concurrents, et ses prédictions étaient comparables aux résultats expérimentaux existants déterminés par des techniques de référence comme la cristallographie par diffraction des rayons X et la microscopie cryo-électronique. Bientôt, je m’attends à ce qu’AlphaFold2 et ses descendants soient les méthodes de choix pour déterminer les structures des protéines avant de recourir à des techniques expérimentales qui nécessitent un travail minutieux et laborieux sur des instruments coûteux.
L’une des raisons du succès d’AlphaFold2 est qu’il a pu utiliser la base de données des protéines, qui compte plus de 170 000 structures 3D déterminées expérimentalement, pour s’entraîner à calculer les structures correctement repliées des protéines.
L’impact potentiel d’AlphaFold peut être apprécié si l’on compare le nombre de toutes les structures protéiques publiées – environ 170 000 – avec les 180 millions de séquences d’ADN et de protéines déposées dans la base de données universelle des protéines. AlphaFold nous aidera à trier les trésors de séquences d’ADN à la chasse de nouvelles protéines aux structures et fonctions uniques.
Est-ce qu’AlphaFold m’a rendu, moi, un modélisateur moléculaire, superflu ?
Comme pour les programmes d’échecs et de Go – AlphaZero et AlphaGo – nous ne savons pas exactement ce que fait l’algorithme d’AlphaFold2 et pourquoi il utilise certaines corrélations, mais nous savons qu’il fonctionne.
En plus de nous aider à prédire les structures de protéines importantes, la compréhension de la « pensée » d’AlphaFold nous aidera également à acquérir de nouvelles connaissances sur le mécanisme de repliement des protéines.
L’une des craintes les plus courantes exprimées à propos de l’IA est qu’elle conduise au chômage à grande échelle. AlphaFold a encore un chemin important à parcourir avant de pouvoir prédire de manière cohérente et avec succès le repliement des protéines.
Cependant, une fois qu’il sera arrivé à maturité et que le programme pourra simuler le repliement des protéines, les chimistes computationnels seront intégralement impliqués dans l’amélioration des programmes, en essayant de comprendre les corrélations sous-jacentes utilisées, et en appliquant le programme pour résoudre des problèmes importants tels que le mauvais repliement des protéines associé à de nombreuses maladies comme Alzheimer, Parkinson, la mucoviscidose et la maladie de Huntington.
AlphaFold et sa progéniture vont certainement changer la façon dont les chimistes computationnels travaillent, mais ils ne seront pas pour autant superflus. D’autres domaines n’auront pas autant de chance. Par le passé, les robots ont pu remplacer les humains effectuant des travaux manuels ; avec l’IA, nos capacités cognitives sont également mises à l’épreuve.
Les robots sont en train d’être mis au défi.