1) Wprowadzenie
Droga odpływu serca (OFT) jest przejściową strukturą na tętniczym biegunie serca, łączącą komory zarodka z workiem aortalnym. OFT formuje się podczas zapętlania się serca z komórek progenitorowych serca w mezodermie gardłowej. Przy maksymalnym wydłużeniu OFT jest skręconym cylindrem mięśnia sercowego wyścielonym komórkami wsierdzia, z ortogonalnymi, ale przylegającymi do siebie domenami proksymalną (conal) i dystalną (truncal). W trakcie tego procesu ściana OFT obraca się i dzieli, tworząc podstawę aorty wstępującej i pnia płucnego. Remodelowanie OFT jest związane z tworzeniem przegrody międzykomorowej, morfogenezą zastawki półksiężycowatej, arteriogenezą naczyń wieńcowych oraz dojrzewaniem mięśnia sercowego i mięśni gładkich podstawy wielkich tętnic. Genetyczne lub środowiskowe zaburzenia elongacji i remodelingu OFT prowadzą do powstania wrodzonych wad serca typu conotruncal, stanowiących 30% wszystkich wrodzonych wad serca (około 0,3% żywych urodzeń) (Bruneau, 2008; Srivastava i Olson, 2000). Szeroko zakrojone badania anatomiczne, eksperymentalne i genetyczne morfogenezy OFT ujawniły udział wielu typów komórek i szlaków sygnalizacyjnych w rozwoju OFT, co sugeruje, że złożoność rozwojowa leży u podstaw zwiększonej częstości występowania anomalii OFT u człowieka.
2) Powstawanie OFT
Podczas wydłużania się cewy sercowej, mięsień sercowy jest stopniowo dodawany do tętniczego bieguna serca z populacji komórek progenitorowych w mezodermie gardłowej określanej jako drugie pole sercowe (SHF; Figura 1A) (Buckingham i in., 2005; Kelly i Buckingham, 2002), (Cai i in., 2003). SHF jest umiejscowione w mezodermie przyśrodkowo, ale przylegająco do komórek dających początek wczesnemu przewodowi sercowemu; następnie komórki SHF leżą pod endodermą brzuszną przedniego jelita, tworząc grzbietową ścianę jamy osierdzia. SHF przyczynia się do powstania prawej komory serca, a także bieguna żylnego i obszarów AV serca; populacja komórek progenitorowych, które przyczyniają się wyłącznie do powstania bieguna tętniczego, znana jest jako przednie lub wtórne pole serca. Zidentyfikowano krytyczne regulatory transkrypcyjne rozwoju SHF, do których należą czynniki transkrypcyjne Isl1, Foxh1, Tbx1, Pitx2 i Mef2c (przegląd w (Buckingham i in., 2005)). Kluczowe właściwości SHF obejmują jego przyśrodkowe położenie, opóźnienie różnicowania i zwiększoną proliferację w stosunku do komórek, które dają początek wczesnemu kanalikowi sercowemu. Właściwości te są regulowane zarówno wewnętrznie, jak i przez otaczające komórki CNC oraz ektodermę i endodermę gardła poprzez aktywność cząsteczek sygnalizacyjnych szlaków czynnika wzrostu fibroblastów, białka morfogenetycznego kości, Hedgehog i Wnt (Cohen i in., 2008; Goddeeris i in., 2007; Park i in., 2008; Prall i in., 2007). Sygnały te wspólnie definiują niszę utrzymującą komórki progenitorowe SHF i regulującą ich stopniowe rozmieszczanie podczas poszerzania OFT (Rochais i in., 2009). OFT jest wyścielony komórkami wsierdzia, które również wywodzą się z mezodermy gardłowej (Laugwitz i in., 2008).
Podczas wydłużania się cewy sercowej, miokardium jest stopniowo dodawane do tętniczego bieguna serca z populacji komórek progenitorowych w mezodermie gardłowej, określanej jako drugie pole sercowe (SHF; Rycina 1A) (Buckingham i in., 2005; Kelly i Buckingham, 2002), (Cai i in., 2003). SHF jest umiejscowione w mezodermie przyśrodkowo, ale przylegająco do komórek dających początek wczesnemu przewodowi sercowemu; następnie komórki SHF leżą pod endodermą brzuszną przedniego jelita, tworząc grzbietową ścianę jamy osierdzia. SHF przyczynia się do powstania prawej komory serca, a także bieguna żylnego i obszarów AV serca; populacja komórek progenitorowych, które przyczyniają się wyłącznie do powstania bieguna tętniczego, znana jest jako przednie lub wtórne pole serca. Zidentyfikowano krytyczne regulatory transkrypcyjne rozwoju SHF, do których należą czynniki transkrypcyjne Isl1, Foxh1, Tbx1, Pitx2 i Mef2c (przegląd w (Buckingham i in., 2005)). Kluczowe właściwości SHF obejmują jego przyśrodkowe położenie, opóźnienie różnicowania i zwiększoną proliferację w stosunku do komórek, które dają początek wczesnemu kanalikowi sercowemu. Właściwości te są regulowane zarówno wewnętrznie, jak i przez otaczające komórki CNC oraz ektodermę i endodermę gardła poprzez aktywność cząsteczek sygnalizacyjnych szlaków czynnika wzrostu fibroblastów, białka morfogenetycznego kości, Hedgehog i Wnt (Cohen i in., 2008; Goddeeris i in., 2007; Park i in., 2008; Prall i in., 2007). Sygnały te łącznie definiują niszę utrzymującą komórki progenitorowe SHF i regulującą ich stopniowe rozmieszczanie podczas poszerzania OFT (Rochais i in., 2009). OFT jest wyścielony przez komórki wsierdzia, które również wywodzą się z mezodermy gardłowej (Laugwitz i in., 2008).
Podczas formowania OFT biegun tętniczy serca jest przesuwany kuudalnie w obszarze gardła, ponieważ zachodzi morfogeneza łuku gardłowego i formowanie obustronnych tętnic łuku aorty (Waldo i in., 2005b). Komórki CNC, wywodzące się z grzbietowego cewy nerwowej, migrują przez tylne łuki gardłowe do OFT serca podczas formowania się dystalnego regionu OFT (Rysunek 1B) (Hutson i Kirby, 2003). Poza krytyczną rolą w przebudowie OFT, komórki CNC odgrywają wczesną rolę w ograniczaniu proliferacji i kontrolowaniu rozmieszczenia SHF (Waldo i in., 2005a). W miarę jak komórki CNC wnikają do dystalnego OFT, komórki śródbłonka w proksymalnym regionie OFT przechodzą proces transformacji nabłonkowej w mezenchymalną w odpowiedzi na sygnały pochodzące z mięśnia sercowego, tworząc, wraz z inwazyjnymi komórkami grzebienia nerwowego, poduszki OFT pomiędzy warstwami mięśnia sercowego i śródbłonka (Sugishita i in., 2004b). Zbieganie się poduszek OFT w strukturę spiralną oddziela przepływ laminarny z embrionalnej lewej i prawej komory. Rozwój poduszeczek zależy od wielu międzykomórkowych szlaków sygnałowych, w tym transformującego czynnika wzrostu b, czynnika wzrostu fibroblastów, białka morpogenetycznego kości, VEGF, Notch i niekanonicznej sygnalizacji Wnt (Wagner i Siddiqui, 2007). OFT osiąga swoją maksymalną długość w połowie ciąży u myszy (E10.5) lub w 4 tygodniu rozwoju u człowieka.
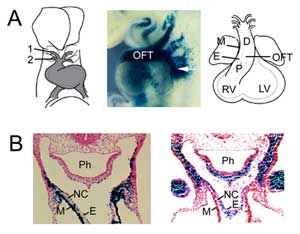
Rysunek 1
Powstawanie OFT. (A) OFT w dniu 8.5 mysiego embrionu jest połączony z pierwszą (1) i drugą (2) tętnicą łuku aorty (po lewej) i rozszerza się przez dodanie komórek progenitorowych drugiego pola serca z mezodermy gardłowej (pośrodku, strzałka), uwidocznione z transgenem pułapki enhancera Fgf10 w E9.5. OFT osiąga maksymalną długość w 10,5 dniu życia embrionalnego myszy i składa się z regionów proksymalnego (P) i dystalnego (D). (B) Porównanie ekspresji transgenu Fgf10 w OFT i drugim polu serca (po lewej) z komórkami pochodzącymi z grzebienia nerwowego (po prawej) w dystalnym OFT w E9.5. Prawy panel reprodukowany z Jiang et al., 2001, Development 127:1607-16, za pozwoleniem. M, myocardium; E, endocardium; RV, right ventricle; LV, left ventricle; Ph, pharynx; NC, neural crest derived cells.
3) Przebudowa OFT
Podczas przebudowy OFT lewa komora uzyskuje niezależne ujście, aortę wstępującą, zapewniając krytyczny systemowy przewód dla natlenowanej krwi po urodzeniu. Cylindryczny OFT przekształca się w podstawę aorty wstępującej i pnia płucnego (ujście prawej komory) równocześnie z przegrodą przedsionkowo-komorową i przebudową tętnicy łuku aorty między 10,5 a 14,5 dniem ciąży u myszy. Przebudowa OFT jest złożonym procesem, w którym udział ma zarówno genetyka, jak i hemodynamika (Yashiro i in., 2007). Ustalono krytyczną rolę rozwoju poduszki OFT i komórek CNC w tym procesie (Sugishita i in., 2004b). Proksymalne miokardium OFT zostaje włączone do ujścia komory, a proces inwazyjnej miokardializacji prowadzi do umięśnienia proksymalnej przegrody międzykomorowej (Rana i in., 2007; van den Hoff i in., 2001). Wrastanie przegrody aortalno-płucnej pomiędzy 4. i 6. tętnicę łuku aorty powoduje rozdzielenie dystalnej części OFT na aortę wstępującą i pień płucny (ryc. 2A). Ściany mięśni gładkich rozwijają się u podstawy wielkich tętnic z końcowego wkładu drugiego pola serca i bardziej dystalnie z CNCs (Sugishita i in., 2004b; Waldo i in., 2005b).
Przegrodom OFT towarzyszy rotacja ściany OFT w kierunku przeciwnym do ruchu wskazówek zegara, co powoduje wyrównanie aorty z lewą komorą i pnia płucnego z prawą komorą (Bajolle i in., 2006). Proces septacji jest zakończony zbieganiem się przegrody międzykomorowej, przedsionkowo-komorowej i międzykomorowej (ryc. 2A). Pierwotna dystalna ściana mięśnia sercowego OFT częściowo współtworzy miokardium podprzeponowe prawej komory, a częściowo ulega programowanej śmierci komórek pod wpływem hipoksji podczas wyrównywania komorowo-tętniczego (Rana i wsp., 2007; Sugishita i wsp., 2004a). Podczas procesu przebudowy w kątowym połączeniu proksymalnych i dystalnych obszarów OFT z pierwotnej i dwóch interkalowanych poduszeczek OFT tworzą się zastawki półksiężycowate (Anderson i in., 2003). Arteriogeneza wieńcowa jest procesem, w którym pochodzący z nasierdzia splot wieńcowy wybiórczo wnika do podstawy aorty powyżej dwóch płatków zastawki skierowanych w stronę pnia płucnego, tworząc ostia łączące układ wieńcowy z aortą wstępującą (Rycina 2B) (Tomanek, 2005).
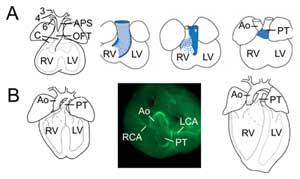
Rysunek 2. emodelowanie dróg odpływu. (A) W dniu embrionalnym 11.5 OFT jest połączony z trzecią (3), czwartą (4) i szóstą (6) tętnicą łuku aorty (po lewej). Przegroda aortalno-płucna (APS) rozdziela dystalny odcinek OFT na aortę wstępującą (Ao) i pień płucny (PT) zbiegające się z poduszkami OFT (C). Prawe trzy obrazy ukazują postępującą rotację ściany mięśnia sercowego podczas przegrody OFT: ściana grzbietowa w dniu embrionalnym 10,5 (niebieski) daje początek brzusznej części mięśnia podprzeponowego. (B) W 14,5 dniu życia zarodka przegroda OFT jest zakończona i aorta jest połączona z lewą komorą, a pień płucny z prawą komorą. W widoku z góry (pośrodku) immunochemia aktyny mięśni gładkich ujawnia połączenia lewej (LCA) i prawej (RCA) tętnicy wieńcowej z podstawą aorty. Struktura ostatecznego serca (po prawej). RV, prawa komora; LV, lewa komora.
4) Wrodzone konotrunalne wady serca
Złożoność rozwoju OFT znajduje odzwierciedlenie w wysokiej częstości występowania anomalii konotrunalnych u człowieka (Bruneau, 2008; Srivastava i Olson, 2000). Niepowodzenia we wprowadzaniu SHF lub CNC lub w przebudowie OFT przyczyniają się do spektrum wad conotruncal obserwowanych w patologii u ludzi i w modelach zwierzęcych (Rycina 3) (Moon, 2008).
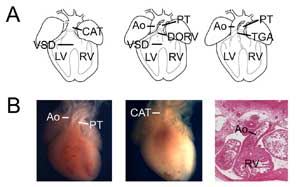
Rycina 3.
Wrodzone wady serca. (A) Karykatury przedstawiające wspólny pień tętniczy (lub przetrwały przewód tętniczy (persistant truncus arteriosus, CAT, po lewej), podwójne ujście prawej komory (double outlet right ventricle, DORV, pośrodku) i przełożenie wielkich pni tętniczych (transposition of the great arteries, TGA, po prawej). VSD, ventricular septal defect; RV, right ventricle; LV, left ventricle, Ao, aorta; PT, pulmonary trunk. (B) Przykłady mysich modeli wrodzonych wad serca. Serce kontrolne ukazujące aortę i pień płucny (po lewej) w porównaniu do serca Tbx1 null (pośrodku) z pojedynczym ujściem komorowym (CAT). Przykład podwójnego ujścia prawej komory typu transpozycyjnego ukazujący aortę połączoną z prawą komorą w sercu zerowym Tbx3 (po prawej).
Podczas gdy poważne upośledzenie rozwoju SHF prowadzi do niewydolności wydłużania się przewodów sercowych, łagodniejsze defekty skutkują anomaliami ułożenia komorowo-tętniczego i brakiem niezależnego połączenia aorty z lewą komorą (Abu-Issa i Kirby, 2007). Te defekty mogą być wewnętrzne dla SHF lub wpływać na otaczające typy komórek (CNC lub nabłonek gardła), które kontrolują niszę komórek progenitorowych SHF. Takie defekty mogą skutkować podwójnym ujściem prawej komory, aortą nadmostkową, hipoplazją płuc, atrezją płucną i tetralogią Fallota. Utrata lub redukcja CNC prowadzi do niepowodzenia lub opóźnienia przegrody OFT, co skutkuje przetrwałym truncus arteriosus lub podwójnym ujściem prawej komory; niedobór CNC wpływa również pośrednio na rozwój SHF, skutkując złożonym fenotypem ułożenia i przegrody (Hutson i Kirby, 2003). Niepowodzenie rotacji ściany OFT podczas przebudowy OFT również skutkuje wadami ułożenia, w tym przełożeniem wielkich tętnic, podwójnym ujściem prawej komory i nadbiegającą aortą, zwykle w następstwie defektów lateralności zarodka (Bajolle i in., 2006; Bamforth i in., 2001). Defekty w tworzeniu przegrody OFT mogą być również wynikiem późniejszego niepowodzenia w różnicowaniu się ściany mięśnia sercowego i nieprawidłowości w procesach istotnych dla przebudowy, takich jak programowana śmierć komórki lub miokardializacja (Park i in., 2008). Wykazano również, że wewnętrzne defekty komórek wsierdzia OFT, jak również zmiany hemodynamiczne, prowadzą do anomalii OFT 16, (Zhang i wsp., 2009), (Bartman i Hove, 2005; Yashiro i wsp., 2007). Wreszcie, nieprawidłowe ułożenie proksymalnych tętnic wieńcowych jest często związane z wadami conotruncalnymi i, w odosobnieniu, stanowi istotną przyczynę nagłej śmierci sercowej (Angelini i in., 2002). Wśród obecnych celów badawczych jest lepsze zrozumienie etiologii wad conotruncal poprzez identyfikację powodujących je mutacji genetycznych i czynników modyfikujących za pomocą ekranów genomowych oraz rozbiór względnego udziału genetycznych i epigenetycznych graczy w tworzeniu i przebudowie OFT przy użyciu modeli zwierzęcych. Badania te dostarczą wglądu w szlaki sygnalizacyjne i procesy komórkowe, które napędzają morfogenezę OFT i są celem mechanizmów chorobowych.
Podziękowania
.