Mundo inteiro, 36 milhões de pessoas têm perda total de visão1. Elas não conseguem ver formas ou mesmo fontes de luz. Para a maioria dessas pessoas, sua cegueira decorre de problemas retificáveis, como cataratas – elas simplesmente não têm acesso a cuidados de saúde adequados. Os milhões restantes, porém, são cegos como resultado de condições que atualmente não têm tratamento eficaz.
“A cegueira é uma das condições que mais altera a vida de uma pessoa”, diz William Hauswirth, o oftalmologista da Universidade da Flórida, em Gainesville. Além das dificuldades que causa para a mobilidade e para encontrar emprego, a deficiência visual está associada a uma série de outros problemas de saúde, incluindo insônia, ansiedade e depressão, e até mesmo o risco de suicídio. “Restaurar a visão útil faria uma melhoria quase inimaginável na qualidade de vida”, diz Hauswirth.
Em países de alta renda onde as causas evitáveis da deficiência visual são tratadas rotineiramente, a principal causa da cegueira é a degeneração da retina. Encontrado na parte de trás do olho, este tecido contém células especializadas que reagem à luz e processam sinais visuais, sendo, portanto, crucial para a visão. As células fotorreceptoras – neurônios comumente conhecidos como hastes e cones – convertem a luz que atinge a retina em sinais eletroquímicos. Estes sinais então filtram através de uma rede complexa de outros neurônios, incluindo células bipolares, células amcrinas e células horizontais, antes de alcançar neurônios conhecidos como células ganglionares da retina. As projeções longas, ou axônios, dessas células formam o nervo óptico, ao longo do qual os sinais da retina são levados até o córtex visual do cérebro, onde são interpretados como imagens.
Desordens retinianas geralmente envolvem a perda de células fotoreceptoras, o que esgota a sensibilidade do olho à luz. Em alguns distúrbios da retina, incluindo a degeneração macular relacionada à idade (DMRI), esta perda resulta da falência das células epiteliais que formam uma camada na parte de trás da retina conhecida como epitélio do pigmento retiniano (EPI). O RPE mantém as células fotorreceptoras saudáveis através da limpeza dos subprodutos tóxicos produzidos durante a reacção com luz, bem como através do fornecimento de nutrientes. Nos distúrbios da retina em que os fotorreceptores permanecem em boa forma, a principal causa da cegueira é a degeneração das células ganglionares da retina.
Oiça uma versão de áudio deste artigo
O seu navegador não suporta o elemento de áudio.
Variedade nas causas da deficiência visual torna mais difícil encontrar soluções. Mas os avanços em várias áreas estão criando esperanças de que quase todas as formas de distúrbio da retina possam se tornar tratáveis.
Uma abordagem é aumentar ou contornar os olhos danificados com próteses funcionais. Esses olhos biônicos podem restaurar apenas uma visão limitada no momento, mas os pesquisadores continuam a pressionar as capacidades dos dispositivos. Outra opção é a terapia genética. Já disponível para pessoas com mutações genéticas específicas, os pesquisadores estão procurando estender esta abordagem a mais pessoas e condições. Alguns cientistas também estão buscando tratamentos baseados em uma técnica relacionada conhecida como optogenética, que envolve a alteração genética das células para restaurar a sensibilidade à luz da retina. Este trabalho está numa fase inicial, mas os investigadores esperam que a abordagem seja capaz de ajudar uma vasta gama de pessoas, porque é agnóstica às causas da degeneração da retina. E os esforços para substituir as células perdidas ou danificadas da retina, seja in situ ou através de transplantes celulares, sugerem que mesmo os distúrbios da retina em estágio tardio podem eventualmente tornar-se tratáveis.
Muito desta pesquisa está na sua infância. Mas Hauswirth está otimista com o progresso que já foi feito. Há dez anos, diz ele, muitas vezes ele tinha que dizer aos pacientes que não podia fazer nada por eles. “Para muitas destas doenças, isso mudou completamente”
Olhos biónicos
Almost 30 anos atrás, Mark Humayun, um engenheiro biomédico da Universidade do Sul da Califórnia em Los Angeles, começou a estimular eletricamente as retinas das pessoas com cegueira. Trabalhando com colegas da Second Sight Medical Products, uma empresa de tecnologia médica em Sylmar, Califórnia, seus experimentos mostraram que tal estimulação poderia induzir a percepção visual de manchas de luz chamadas fosfenos. Depois de uma década de trabalho em animais para estabelecer a quantidade de corrente elétrica que poderia ser aplicada com segurança ao olho, e armado com um conhecimento muito maior sobre o número e os tipos de células que persistem na degeneração das retinas humanas, a equipe de Humayun estava pronta para começar a trabalhar com pessoas. Entre 2002 e 2004, os pesquisadores implantaram um olho biônico em cada uma das seis pessoas que tinham cegueira total ou quase total em um olho – a primeira experiência deste tipo. Os receptores do dispositivo, conhecido como Argus I, relataram ser capazes de perceber fosfenos, movimento direcional e até formas2. Cerca de 300 pessoas experimentam agora o mundo através do sucessor desse dispositivo, o Argus II, que foi aprovado pelos reguladores na Europa em 2011 para uso em pessoas com retinite pigmentosa – um grupo de doenças genéticas raras que causam a degeneração das células fotoreceptoras. A US Food and Drug Administration (FDA) seguiu o exemplo dois anos depois.

O implante Argus II é composto por um conjunto de eléctrodos que está ligado à superfície da retina.Crédito: Ringo Chiu/ZUMA /Alamy
p> Para ser equipado com um Argus II, os pacientes são submetidos a cirurgia para fixar um chip contendo um conjunto de eléctrodos à superfície da retina. Para “ver” com o dispositivo, uma câmara de vídeo miniatura montada num par de relés de óculos sinaliza para uma unidade de processamento que é usada pelo receptor. O processador converte os sinais em instruções que são transmitidas sem fio para o dispositivo implantado. Os eletrodos estimulam então as células ganglionares da retina na parte frontal da retina. A utilização da prótese é um processo de aprendizagem. Os receptores devem treinar o seu cérebro para interpretar o novo tipo de informação recebida. E como a câmera de vídeo não acompanha o movimento do olho, eles também devem aprender a mover a cabeça para dirigir o olhar.p>O dispositivo fornece apenas visão limitada. Os usuários podem detectar fontes de luz e objetos com bordas de alto contraste, tais como portas ou janelas, e alguns podem decifrar letras grandes. Estas limitações surgem em parte porque os 60 eléctrodos do dispositivo proporcionam uma resolução muito baixa em comparação com os milhões de células fotorreceptoras de um olho saudável. Mas mesmo este melhoramento mínimo pode melhorar consideravelmente a vida das pessoas.
Onde o Argus II é um implante epiretinal – o que significa que se encontra na superfície da retina – outros dispositivos em desenvolvimento são concebidos para serem colocados por baixo da retina. Estes implantes subretinais podem estimular células que estão mais próximas daquelas que normalmente introduzem sinais na retina – as células fotorreceptoras. Ao estimular as células mais acima na via visual, os investigadores esperam preservar mais do processamento do sinal que é realizado por uma retina saudável.
Retina Implant, uma empresa de biotecnologia com sede em Reutlingen, Alemanha, construiu um implante subretinal composto por fotodíodos (dispositivos semicondutores que convertem luz em corrente eléctrica) que sentem directamente a luz a entrar no olho. Isto elimina a necessidade de uma câmara de vídeo externa, permitindo aos utilizadores dirigir o seu olhar naturalmente. A energia é fornecida por uma unidade portátil, através de uma bobina que é implantada sob a pele, acima da orelha. Alpha AMS, a versão atual do sistema, recebeu aprovação regulamentar na Europa para uso em pessoas com retinite pigmentosa.
Pixium Vision em Paris está testando um implante subretinal fotovoltaico chamado Prima. O sistema projecta sinais de uma câmara de vídeo montada em óculos para o olho utilizando luz infravermelha próxima, cujo comprimento de onda acciona de forma óptima os fotodíodos no dispositivo para estimular as células da retina. Projetar imagens desta forma dá aos usuários algum controle sobre a direção do seu olhar, pois eles podem explorar a cena movendo apenas seus olhos. A energia também é fornecida pela luz infravermelha próxima, tornando o implante sem fio e a cirurgia para encaixá-lo menos complicada. “Os pacientes estão aprendendo a recuperar a visão mais rapidamente, e a resolução parece melhor”, diz José-Alain Sahel, oftalmologista da Universidade de Pittsburgh, Pensilvânia, que está conduzindo testes de segurança do dispositivo em dez pessoas com AMD. “É cedo, mas isto é muito promissor”
Todos estes dispositivos só funcionam quando as células em funcionamento permanecem na retina. Em condições oculares comuns que afetam principalmente as células fotorreceptoras, incluindo a retinite pigmentosa e a DMRI, geralmente restam algumas células para estimular. Mas quando muitas células ganglionares da retina morrem, como ocorre na retinopatia diabética avançada e no glaucoma, tais implantes não podem ajudar. Para pessoas sem nenhuma função retiniana remanescente, seja devido a doença ou lesão, uma abordagem biônica alternativa pode ser mais relevante.
Humayun e seus colegas estão trabalhando em um sistema que contorna o olho enviando sinais diretamente para o cérebro. A idéia não é nova: nos anos 70, o engenheiro biomédico americano William Dobelle mostrou que estimular diretamente o córtex visual desencadeou a percepção dos fosfenos3. Mas a tecnologia dos olhos biónicos só agora está a recuperar o atraso. A Second Sight desenvolveu o Orion, um sistema que é, segundo Humayun, “basicamente um Argus II modificado”. Da mesma forma que o original, ele usa uma câmera de vídeo e um processador de sinal que se comunica sem fio com um implante, mas o chip é colocado na superfície do córtex visual e não na retina. O dispositivo está a ser testado em cinco pessoas com percepção de luz limitada ou nula devido a uma lesão ocular ou danos na retina ou no nervo óptico. “Até agora, os resultados são bons”, diz ele. “Ainda não estamos surpresos com nada”
Dado que parte da tecnologia já está testada e testada em pessoas, Humayun está otimista de que o sistema poderia receber aprovação regulatória dentro de alguns anos. “Obviamente, a cirurgia cerebral tem um nível de risco diferente, mas o procedimento é bastante simples, e o Orion poderia ajudar muito mais pacientes”, diz ele. No entanto, sabe-se muito menos sobre estimular o cérebro para proporcionar uma visão útil. “Sabemos muito sobre a retina, mas muito pouco sobre o córtex”, diz Botond Roska, um neurobiólogo do Instituto de Oftalmologia Molecular e Clínica da Basileia, na Suíça. Mas nunca saberemos o suficiente se não tentarmos”, diz ele.
Terapia genética
O olho é um alvo ideal para a terapia genética”. Por ser relativamente independente, os vírus que são usados para transportar genes para as células da retina não devem ser capazes de viajar para outras partes do corpo. E porque o olho é um local imunoprivilegiado, o sistema imunitário tem menos probabilidades de montar uma defesa contra tal vírus.
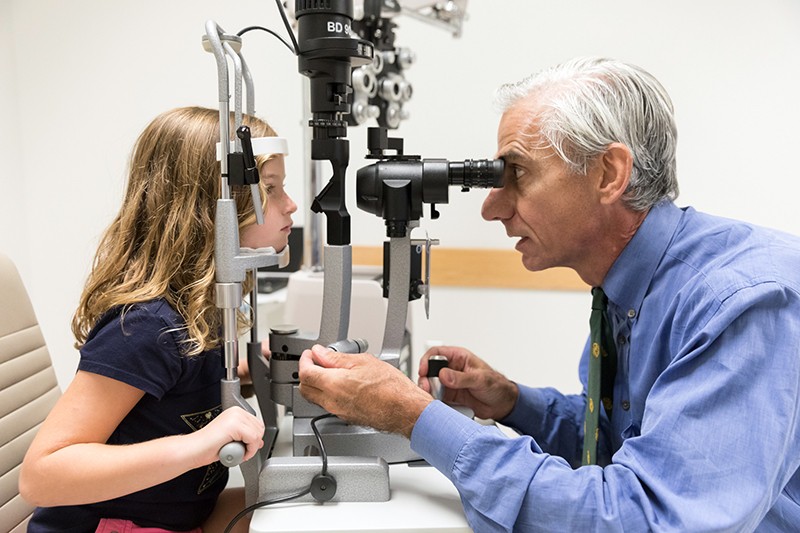
Oftalmologista Albert Maguire examina os olhos de uma menina com amurose congénita de Leber cuja visão foi restaurada pela terapia genética voretigene neparvovec (Luxturna).Crédito: Hospital Infantil de Filadélfia
Na primeira demonstração do potencial da terapia genética para combater a cegueira, três equipes de pesquisadores utilizaram a técnica para tratar com sucesso pessoas com amaurose congênita de Leber (LCA). Esta condição hereditária leva a uma grave deficiência visual e começa nos primeiros anos de vida, manifestando-se frequentemente como cegueira nocturna antes de progredir para uma perda ampla da visão que começa na periferia do campo visual. Ela afeta cerca de 1 em 40.000 bebês.
Os pesquisadores se propuseram a enfrentar uma forma específica da condição conhecida como LCA 2. Isto é causado por mutações na RPE65, um gene que é expresso pela RPE. O gene mutado afeta negativamente a função RPE, que por sua vez danifica as células fotoreceptoras. Em 2008, as três equipes, incluindo uma liderada por Hauswirth, mostraram em ensaios clínicos em estágio inicial que a entrega de uma cópia saudável da RPE65 à retina era segura e levou a melhorias limitadas na visão4,5,6. Um ensaio clínico fase III liderado por Albert Maguire, oftalmologista da Universidade da Pensilvânia na Filadélfia, mostrou em Agosto de 2017 que as pessoas com LCA 2 que receberam o tratamento eram mais capazes de navegar em percursos de obstáculos a vários níveis de iluminação do que as que não o fizeram7. Em dezembro de 2017, a FDA aprovou o tratamento, voretigene neparvovec (Luxturna), tornando-o a primeira terapia genética para qualquer condição a obter luz verde para uso clínico.
É possível tratar o LCA 2 desta forma porque as mutações genéticas envolvidas mostram um padrão recessivo de herança. Isto significa que ambas as cópias de uma pessoa de RPE65 devem carregar as mutações relevantes para causar o distúrbio. O fornecimento de uma única versão, não mutada, resolve o problema. Condições que são causadas por mutações herdadas de forma dominante, entretanto, requerem apenas uma cópia mutante de um gene para se manifestar. Na maioria delas, a simples adição de uma cópia normal do gene não ajudará; em vez disso, o gene mutado deve ser inativado. Uma opção é silenciá-la adicionando moléculas específicas de RNA que interceptam as instruções do gene mutado para fazer a proteína defeituosa, e então fornecer uma cópia normal do gene para assumir suas funções – uma abordagem chamada supressão e substituição. Outra é corrigir a mutação usando a técnica de edição de genes CRISPR-Cas9. Pesquisadores da Universidade de Modena e Reggio Emilia em Modena, Itália, demonstraram esta abordagem em um modelo de retinite pigmentosa8 de rato em 2016. No ano seguinte, uma equipe nos Estados Unidos a utilizou para corrigir a mutação que causa um tipo de glaucoma tanto em ratos quanto em células humanas cultivadas9,
Um importante fator de progresso da terapia genética tem sido o uso do vírus adeno-associado (AAV) para fornecer genes substitutos às células. Os AAV demonstraram ser seguros, em parte, porque tendem a não se integrar no genoma da célula hospedeira, o que minimiza o risco de as células se tornarem cancerosas. E o seu pequeno tamanho permite-lhes uma ampla difusão através do olho e, portanto, infectar um grande número de células. Mas a capacidade dos AAV de fornecer genes tem limites: alguns genes são simplesmente grandes demais para que os AAV carreguem, incluindo o ABCA4, mutações nas quais podem levar à doença de Stargardt, uma forma herdada de degeneração macular. Duas soluções estão sendo seguidas. A primeira utiliza um vírus com maior capacidade de transporte, como um lentivírus, para fornecer genes substitutos. A segurança e eficácia desta abordagem é desconhecida, mas os ensaios clínicos estão em curso. Uma segunda estratégia é quebrar o gene substituto em dois e transportar cada metade separadamente para dentro da célula, juntamente com um meio de recombiná-los. “Isso está funcionando em pelo menos um modelo animal agora”, diz Hauswirth.
Independentemente da abordagem, a terapia genética tem uma limitação considerável. Mais de 250 genes estão implicados na cegueira, e como cada um pode ser afetado por inúmeros tipos de mutação, o número de alvos terapêuticos potenciais é enorme. Por exemplo, mais de 100 mutações no gene RHO levam à retinite pigmentosa, o mais comum distúrbio predominantemente herdado da retina. Desenvolver uma terapia genética para cada mutação não é prático, diz Hauswirth.
P>Pesquisadores estão trabalhando em uma solução potencial que coloca uma reviravolta na abordagem de supressão e substituição. Ao invés de visar cópias de RHO contendo uma mutação específica, eles usam um RNA silencioso para suprimir toda expressão do gene, seja RHO mutado ou não, enquanto entregam uma cópia substituta que é imune ao RNA silencioso. Uma equipe liderada por Jane Farrar, geneticista do Trinity College, Dublin, mostrou a promessa desta estratégia em 2011 em um modelo de retinite pigmentosa10 dominante no rato. Em 2018, Hauswirth e colegas testaram a abordagem em cães com retinite pigmentosa11. Eles mostraram que a degeneração de células fotorreceptoras em áreas tratadas da retina poderia ser interrompida – uma melhora que persistiu por pelo menos oito meses. Esta estratégia trata todas as mutações que podem causar retinite pigmentosa hereditária dominante em um único tratamento e, portanto, estende a terapia genética de condições recessivas para condições hereditárias dominantes “de uma maneira bastante simples”, diz Hauswirth. Ele planeja estudar como cães que receberam o tratamento podem navegar num labirinto, e está coletando os dados de segurança necessários para iniciar um ensaio clínico.
Optogenética
A terapia genética funciona apenas em pessoas cuja cegueira é causada por mutação genética. Também não é apropriada para combater a doença retiniana em fase terminal, na qual permanece um número insuficiente de células a serem reparadas. Mas uma abordagem relacionada baseada em uma técnica chamada optogenética é o agnóstico da doença e pode levar a tratamentos para diferentes estágios de degeneração. Na optogenética, os genes que permitem que as células produzam proteínas sensíveis à luz, conhecidas como opsinas, são fornecidos por um vírus. A introdução de opsinas pode restaurar alguma sensibilidade à luz para os fotorreceptores danificados, ou mesmo fazer outras células da retina, incluindo células bipolares ou células ganglionares da retina, sensíveis à luz.

Optogenética tem sido usada para restaurar a sensibilidade à luz das células cones (verdes) em um modelo de retinite pigmentosa do rato; o sucesso da técnica foi avaliado medindo a atividade de uma célula ganglionar da retina (magenta), que é estimulada pelos cones em resposta à luz.Crédito: IOB.ch
Problematicamente, entretanto, enquanto as células fotoreceptoras do olho podem lidar com uma ampla gama de intensidades de luz – trabalhando bem tanto na luz solar brilhante quanto no crepúsculo – as opsinas têm um alcance limitado e muitas vezes apresentam melhor desempenho em altas intensidades de luz. Uma solução potencial é utilizar uma configuração que funcione de forma semelhante ao sistema Prima bionic-eye da Pixium Vision, no qual os receptores são equipados com óculos que incorporam uma câmara de vídeo que captura a visão do utilizador e um projector que aponta para o seu olho. Como no Prima, o benefício é que a natureza da luz que entra no olho pode ser adaptada à modificação da retina; no entanto, neste caso, a intensidade e o comprimento de onda escolhidos são os que melhor impulsionam as opsinas recém introduzidas em vez de fotodiodos implantados.
GenSight Biologics, uma empresa de biotecnologia em Paris que conta Sahel e Roska entre seus fundadores, já está testando tal sistema. O seu objectivo é fornecer uma opsina às células ganglionares da retina, mas existe um problema potencial: as células ganglionares da retina são naturalmente sensíveis à luz. Elas expressam melanopsina, uma proteína envolvida no reflexo de luz pupilar, na qual a pupila do olho se contrai em resposta à luz brilhante. Para evitar que isso seja desencadeado, os pesquisadores da GenSight estão usando uma opsina que responde aos comprimentos de onda vermelhos da luz, porque a melanopsina responde preferencialmente à luz na extremidade azul do espectro. A empresa iniciou um ensaio clínico em fase inicial em Outubro de 2018 em pessoas com retinite pigmentosa avançada que têm uma visão mínima remanescente. O ensaio envolverá coortes do Reino Unido, França e Estados Unidos, e os resultados iniciais são esperados até o final de 2020.
“Esta é uma abordagem simples, e teremos de ver o que será ganho”, diz Roska. “Então, podemos avançar para abordagens cada vez mais sofisticadas”. Um problema que permanece é que muitas das doenças que as técnicas optogenéticas podem tratar envolvem a degeneração de partes específicas da retina, sendo a visão útil retida em outras áreas. A luz que conduz as opsinas é visível e pode interferir com a visão natural remanescente. No futuro, as opsinas que respondem à luz infravermelha próxima poderão permitir que os tratamentos optogenéticos funcionem em conjunto com a visão natural residual.
Regeneração de células
Terapia com células estaminais poderá potencialmente curar a cegueira mesmo nas fases tardias da doença. Como as células estaminais podem ser coaxadas para se tornarem qualquer tipo de célula, elas poderiam ser usadas para cultivar células retinianas frescas para transplante para o olho para substituir as que foram perdidas. Contudo, estudos em animais mostraram que apenas uma pequena proporção dos neurónios transplantados é capaz de se integrar correctamente no complexo circuito neurológico da retina. Este é um obstáculo considerável para os tratamentos com células estaminais que visam substituir os neurónios da retina.

A estrutura celular complexa da retina inclui camadas de fotorreceptores (verdes) e vasos sanguíneos e nervos (magenta).Crédito: Louise Hughes/SPL
As células que compõem o epitélio pigmentar da retina, por outro lado, ficam fora dos circuitos da retina. As terapias baseadas em células estaminais são, portanto, as mais promissoras para doenças, como a DMRI e a retinite pigmentosa, que causam a degeneração das células RPE. “Os fotorreceptores têm de se ligar aos circuitos, mas o epitélio do pigmento retiniano não”, diz Roska. “É aí que as pessoas estão mais perto de fazer progressos.” Inicialmente, os pesquisadores tentaram injetar a retina com células RPE derivadas de células-tronco em suspensão, mas muito poucas ficaram presas onde eram necessárias. Várias equipas pensam agora que uma melhor abordagem é transplantar células RPE para o olho como uma folha pré-formada que é depois mantida em posição por um andaime biocompatível. “A abordagem do andaime é uma enorme melhoria, em comparação com a suspensão, para as células RPE”, diz Sahel.
Em março de 2018, o London Project to Cure Blindness – uma colaboração entre o University College London e o Moorfields Eye Hospital em Londres – anunciou os resultados de um ensaio fase I no qual uma folha de células RPE foi implantada na retina de duas pessoas com DMRI úmida (uma forma rara e séria de DMRI envolvendo crescimento anormal e vazamento de vasos sanguíneos). Ambos os receptores toleraram bem o procedimento e foram capazes de ler 21-29 mais cartas em um gráfico de leitura do que antes do tratamento12. No mês seguinte, uma equipe liderada por Humayun relatou que a fase I similar resultou em cinco pessoas com DMRI seca, a forma mais comum da doença13. Estes resultados iniciais estão cheios de promessas. “Isto levou a muita excitação”, diz Humayun. Mas os resultados precisam ser confirmados por estudos fase III em um número maior de participantes, e Humayun adverte que o tratamento pode estar a muitos anos de ser usado na clínica, porque nenhuma terapia com células-tronco para um distúrbio da retina ainda passou pelo processo de aprovação.
Uma abordagem relacionada, ainda nos estágios iniciais da pesquisa básica, poderia satisfazer a esperança de substituir neurônios perdidos, abrindo a porta para tratamentos para uma grande variedade de doenças oftalmológicas. Em humanos, os neurônios maduros não se dividem e, portanto, não podem se regenerar, o que torna a restauração da visão especialmente difícil. Mas o mesmo não se aplica a todos os animais. Répteis e certos peixes podem regenerar os neurônios da retina, e aves também exibem alguma capacidade regenerativa. Thomas Reh, um neurocientista da Universidade de Washington em Seattle, está tentando desbloquear essa capacidade em humanos. Mas ao invés de transplantar células cultivadas em laboratório, Reh pretende coaxar células que já estão na retina para se diferenciar em neurônios frescos.
Em 2001, Reh sugeriu que a gláia de Müller – células que fornecem estrutura à retina e suportam sua função – são a fonte de novos neurônios que tinham sido observados em peixes e aves14. Ele e sua equipe começaram então a descobrir se Müller glia poderia ser usada para gerar neurônios frescos em camundongos. Em 2015, eles projetaram ratos para fazer Ascl1, uma proteína importante para a produção de neurônios em peixes, e depois danificaram as retinas dos animais15. A esperança deles era que o Ascl1 provocasse a Müller glia a se transformar em neurônios.
A experiência não conseguiu produzir novos neurônios em ratos adultos, mas teve sucesso em ratos jovens. Nikolas Jorstad, um bioquímico e aluno de doutorado da equipe de Rehs, propôs que modificações químicas feitas na cromatina (um complexo de DNA, RNA e proteínas) no núcleo celular durante o desenvolvimento poderiam bloquear o acesso em células maduras a genes que permitem a transformação da gláia de Müller em neurônios. Em agosto de 2017, a equipe de Reh mostrou que ao introduzir uma enzima que reverte tais modificações, eles poderiam coaxar a glia de Müller para se diferenciar16. “Pela primeira vez, poderíamos regenerar neurônios no rato adulto”, diz Reh. “Depois de todos estes anos, fiquei bastante entusiasmado.” Embora não fossem verdadeiras células fotorreceptoras, e se pareciam mais com células bipolares, os neurônios se ligavam aos circuitos existentes, e eram sensíveis à luz. “Fiquei surpreso que eles se conectassem tão bem quanto eles”, diz Reh.
Embora longe de estar pronto para tratar distúrbios da retina nas pessoas, o trabalho tem um enorme potencial. O próximo passo será repetir os estudos em animais com olhos mais parecidos com os humanos. A equipe de Reh já está trabalhando com culturas de células da retina de primatas não humanos. Os pesquisadores também precisam trabalhar em como direcionar o processo de diferenciação para produzir tipos específicos de células, como varas e cones. “Agora que temos o pé no negócio de fabricação de neurônios, os cones seriam ótimos”, diz Reh.
Se bem sucedido, a abordagem poderia ser amplamente aplicável. “Em última análise, esta será a forma como todas estas doenças dos olhos serão tratadas”, prevê Reh. “Apenas faz sentido. Você não tem que se preocupar em fazer os transplantes certos. Suas células estão exatamente onde você precisa delas.”
Humayun também é encorajado pelo trabalho. “Torço por qualquer pessoa com uma nova boa ideia”, diz ele. “É muito cedo, é de alto risco, mas nunca digas nunca. Foi o que eu aprendi.”