- Introduktion
- Material och metoder
- Provberedning
- MinION-biblioteksberedning och sekvensering
- Genomisk analys
- Resultat
- MinION-sekvenseringsdata och sammansättning av virusgenom
- Utvidden och källan till kontaminering mellan prover
- Diskussion
- Författarbidrag
- Finansiering
- Intressekonfliktförklaring
- Acknowledgments
- Supplementary Material
- Footnotes
Introduktion
Metagenomisk sekvensering har potential att möjliggöra opartisk identifiering av patogener från ett kliniskt prov. Den lovar att fungera som en enda och universell analys för diagnostik av infektionssjukdomar direkt från prover utan behov av a priori kunskap (Bibby, 2013; Miller et al., 2013; Schlaberg et al., 2017). Förutom identifiering av patogenarter kan breda och djupa metagenomiska sekvensdata ge information som är relevant för att fastställa behandling och prognos, upptäcka utbrott och spåra infektionsepidemiologi (Greninger et al., 2010; Yang et al., 2011; Qin et al., 2012; Loman et al., 2013). Plattformar för nästa generations sekvensering (NGS) kan producera en massiv genomströmning av data till en blygsam kostnad, men dess tillämpning inom klinisk diagnostik och folkhälsa har begränsats av komplexitet, långsamhet och kapitalinvesteringar.
MinION är en palmstor, realtidsgenomsekvensator i enstaka molekyler i realtid som utvecklats av Oxford Nanopore Technologies (ONT). Minions kompakta storlek och realtidskaraktär skulle kunna underlätta tillämpningen av metagenomisk sekvensering vid testning av infektionssjukdomar på plats i vården, vilket visats av flera proof-of-concept-studier, inklusive identifiering av Chikungunya (CHIKV), Ebola (EBOV) och hepatit C-virus (HCV) från kliniska blodprover från människor utan målberikning (Greninger et al, 2015), och detektion av bakteriella patogener från urinprover (Schmidt et al., 2016) och luftvägsprover, utan behov av föregående odling (Pendleton et al., 2017).
Datagenomströmningen av MinION har ökat kraftigt sedan lanseringen 2015, och varje förbrukningsbar flödescell genererar nu upp till 10-20 Gb DNA-sekvensdata. Detta gör det möjligt för användare att använda flödescellen mer effektivt (och minska kostnaderna) genom att multiplexera flera prover i en enda sekvenseringskörning. ONT har utvecklat PCR-fria streckkodssatser som gör det möjligt att multiplexa upp till 12 prover.
Detektion av influensa A-virus i flera respiratoriska prover skulle kunna vara en diagnostisk användning av en multiplexad MinION-sekvensanalys. När man sekvenserar direkt från prover med ett potentiellt brett spektrum av virala titrar är det dock viktigt att vara medveten om risken för korskontaminering av proverna, både under förberedelserna av biblioteken och det bioinformatiska steget för demultiplexering av streckkoder efter sekvenseringen. Här presenterar vi ett unikt MinION-sekvenseringsdataset och resultaten av en undersökning av omfattningen och källan till korskontaminering av streckkoder vid multiplexsekvensering.
Material och metoder
Vi använde ett nässköljprov från en iller som var infekterat med influensa A-virus som ett exempel och spikade också två alikvots av negativa nässköljprover från icke-infekterade illrar (redan existerande, oanvända lager från en icke-relaterad studie) med dengue- och chikungunyavirus separat. Inget av dessa virus är relevant för klinisk diagnostik i andningsprover, men fungerar här som tydliga, distinkta markörer för bedömning av korskontaminering av prover. Sekvenseringsbiblioteken för varje prov framställdes parallellt, tillsammans med en negativ nässköljningskontroll, barkodades och sekvenserades individuellt. Vi sammanförde sedan en alikvot av sekvenseringsbiblioteken och utförde multiplex MinION-sekvensering. Läsningar från de fyra individuella körningarna (kallade ”CHIKV”, ”DENV”, ”FLU-A” och ”Negativ”) och multiplexkörningen (kallad ”Multiplexed”) analyserades sedan för att undersöka omfattningen och källan till tvärprovsförorening.
Provberedning
Projektlicensen granskades av den lokala AWERB (Animal Welfare and Ethics Review Board) och beviljades därefter av inrikesministeriet. RNA extraherades med hjälp av QIAamp viral RNA-kitet (Qiagen) enligt tillverkarens anvisningar från nässköljning av illrar som innehöll influensa A (H1N1)-virus (A/California/04/2009) och en pool av negativa nässköljningsprover. Alikvots av det negativa provextraktet spikades med antingen denguevirus (DENV) (stam TC861HA, GenBank: MF576311) eller CHIKV (stam S27, GenBank: MF580946.1) viralt RNA från The National collection of Pathogenic Viruses1. Proverna behandlades med DNase med TURBO DNase (Thermo Fisher Scientific, Waltham, MA, USA) och renades med hjälp av RNA Clean & ConcentratorTM-5-kitet (Zymo Research). cDNA framställdes och amplifierades med hjälp av en sekvensoberoende singelprimeramplifieringsmetod (Greninger et al., 2015), modifierad som tidigare beskrivits (Atkinson et al., 2016). Amplifierat cDNA kvantifierades med hjälp av Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA), och 1 μg användes som input för varje MinION-biblioteksberedning, med undantag för den negativa kontrollen där hela provet (32 ng) användes.
MinION-biblioteksberedning och sekvensering
Ligation Sequencing Kit 1D (SQK-LSK108) och Native Barcoding Kit 1D (EXP-NBD103) användes enligt ONT:s standardprotokoll, med undantaget att endast en streckkod ingick i var och en av de fyra bibliotekspreparationerna. Varje bibliotek kördes på en enskild flödescell och ett femte poolat bibliotek gjordes genom att kombinera de fyra individuellt streckkodade biblioteken. Biblioteken sekvenserades på R9.4 flödesceller. Studiens utformning visas i figur 1.
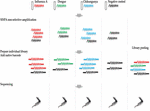
FIGUR 1. Översikt över studiens uppläggning. RNA extraherades från fyra prover, inklusive ett prov från en illers nässköljning som infekterats med influensa A-virus, två negativa prover från en illers nässköljning som spetsats med dengue- och chikungunyavirus och en negativ kontroll från en illers nässköljning. cDNA framställdes och amplifierades med hjälp av en sekvensoberoende enkelprimersamplifieringsmetod. Sekvenseringsbiblioteken för varje prov framställdes parallellt, barkodades och sekvenserades på enskilda flödesceller. Multiplexsekvensering utfördes också genom att de fyra individuella biblioteken sammanfördes. Läsningar från de fyra individuella körningarna och multiplexkörningen analyserades för att bedöma omfattningen och källan till tvärgående streckkodskontaminering vid multiplexsekvensering.
Genomisk analys
Läsningar baskallas med hjälp av Albacore v2.1.7 (ONT) med streckkodsdemultiplexering. Läsningar från varje sekvenseringskörning kartlades till genomiska sekvenser för varje virus med Minimap2 (Li, 2018). Antalet läsningar som mappades till referensen räknades med hjälp av Pysam2. De novo-samlingen utfördes med Canu v1.7 (Koren et al., 2017), och det resulterande utkastet till genomet polerades med Nanopolish (Mongan et al., 2015) med signalnivådata.
För att möjliggöra sträng streckkodsdemultiplexering av Multiplex MinION-sekvenseringsdata utförde vi två analysrundor med Porechop (v0.2.23). Förekomst av en adaptersekvens i mitten av en läsning är en signatur för chimera. Vi använde Porechop för att undersöka varje läsning och de som har en mittregion som delar >75 % identitet med adaptersekvensen identifierades som chimärläsningar. I Porechop ställer vi in alternativet ”-middle_threshold” och väljer ett tröskelvärde på 75. I den andra omgången använde vi Porechop för att söka upp streckkodssekvensen både i början och i slutet av en läsning; läsningar tilldelades endast om samma streckkod hittades i båda ändarna. Vi ställde in alternativet ”-require_two_barcodes” i Porechop och fastställde tröskelvärdet för streckkodsscore till 70. För att hitta potentiella signaturer för chimära läsningar undersökte vi de läsströmssignaler som lagrades i FAST5-filen med MinION-sekvensatorn. Strömsignalerna extraherades med hjälp av ONT fast5 API4 och plottades med hjälp av ggplot2 implementerat i R5 för en jämförelse av chimära och icke-chimära läsningar.
Resultat
MinION-sekvenseringsdata och sammansättning av virusgenom
Genomströmningen för varje MinION-sekvenseringskörning varierade på grund av skillnader i körtid. Ett maximalt antal ∼2,4 M läsningar uppnåddes av den multiplexade sekvenseringskörningen och den individuella CHIKV-körningen, på grund av längre körtider (kompletterande tabell S1). Läsningar från det spikade viruset stod för 96 % av data i de enskilda CHIKV- och DENV-sekvenseringskörningarna och 78 % för FLU-A-provet (tabell 1). Procentandelen virusavläsningar inom varje streckkodat prov i de multiplexade sekvenseringsuppgifterna ligger nära den i de individuellt körda provuppgifterna (tabell 2). Varje virusgenom hade ett extremt högt (>8 000) genomsnittligt täckningsdjup i de individuella och multiplexa sekvenseringsdata, och de novo-sammansättning kunde återskapa nästan fullständiga genomer för alla tre virus med 99.9 % identiteter jämfört med GenBank-referensen.

TABELL 1. Sammanfattning av kartläggnings- och de novo-samlingsresultat för data från MinION-sekvensering av enskilda bibliotek.

TABELL 2. Sammanfattning av kartläggnings- och de novo-sammansättningsresultat för data från multiplex MinION-sekvensering.
Utvidden och källan till kontaminering mellan prover
Varje prov var streckkodat och sekvenserades både individuellt och multiplexerat, vilket gjorde det möjligt för oss att undersöka prestandan av streckkodsdemultiplexering av långfenad tonfisk. I de individuellt sekvenserade provdata skulle vi förvänta oss att endast en enda inhemsk streckkod skulle förekomma. För CHIKV (streckkod NB01), DENV (NB09) och FLU-A (NB10) fann vi att 86, 109 respektive 17 reads tilldelades streckkodsbins som inte förväntades finnas i biblioteket (vilket motsvarar 0,0036, 0,0129 och 0,001 % av de totala reads). I multiplexsekvenseringsdata tilldelades 41 reads (0,0016 %) till streckkoder som inte ingick i experimenten (dvs. en annan streckkod än NB01, NB05, NB09 eller NB10). Vi definierade dessa som felaktigt tilldelade läsningar (figur 2A).
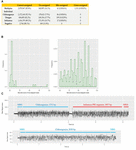
FIGUR 2. (A) Sammanfattning av antalet och procentandelen läsningar som tilldelats korrekt, inte tilldelats, tilldelats felaktigt och korsvis tilldelats i varje sekvenseringskörning. Icke tilldelade refererar till läsningar som Albacore inte kan tilldela någon bins på grund av att streckkodspoängen är mindre än 60. Felaktigt tilldelade refererar till läsningar som tilldelades streckkodsbins som inte ingick i det här försöket och tvärsäkert tilldelade refererar till läsningar som tilldelades de felaktiga streckkodsbins. B) Fördelning av streckkodspoäng som rapporterades av Albacore för felaktigt tilldelade läsningar och tvärsäkert tilldelade läsningar i multiplexsekvensdata. C) Jämförelse mellan den råa signalen från en chimär och en korrekt tilldelad läsning. Signalen för en chimär avläsning har en signal med en stall-signal och en stor spik-signal i mitten av avläsningen.
För att undersöka potentiell laboratoriekontaminering vid beredningen av sekvenseringsbiblioteken kartlade vi alla avläsningar från varje enskild körning mot genomsekvenserna för alla tre virusen. Ingen avläsning visade sig härröra från ett genom som framställts i ett annat bibliotek, vilket tyder på att det inte förekom någon in vitro-kontaminering. Multiplex-sekvenseringsbiblioteket framställdes genom att man sammanförde de enskilda, icke-kontaminerade biblioteken efter ligering av både streckkod och adapter. Kartläggningsresultaten visar dock att 1 311 (0,0543 %) reads kartlades till det felaktiga målgenomet, vilket innebär att de korstilldelades till fel streckkodsbins (senare kallat ”korstilldelade reads”), trots att det multiplexade sekvenseringsbiblioteket var poolat med enskilda bibliotek som inte visade några korstilldelade reads överhuvudtaget. Vi antog att felaktigt tilldelade och korsvis tilldelade läsningar berodde på en låg streckkodspoäng och undersökte streckkodspoängen för dessa läsningar. De flesta av de felaktigt tilldelade läsningarna hade en streckkodspoäng <70, men de korsvis tilldelade läsningarna hade mer varierande poäng, från 60 till nästan 100 (figur 2B). Detta resultat tyder på att felaktigt tilldelade och korsvis tilldelade läsningar kommer från olika källor. Vi analyserade de korstilldelade läsningarna mot en liten databas som omfattar genomsekvenserna av de tre virus som ingick i den här studien och visade att 1074/1311 (82 %) av dessa läsningar kunde korsjusteras mot mer än ett virusgenom (1 047 läsningar) eller korsjusteras mot olika regioner inom samma genom (27 läsningar), vilket tyder på att det rör sig om chimärer. För att bekräfta denna observation undersökte vi de råa strömsignalerna för några korsassocierade läsningar jämfört med dem för korrekt tilldelade läsningar (figur 2C). De aktuella signalerna för en korrekt tilldelad läsning omfattar vanligtvis följande: (i) en signal för öppen pore med hög ström som representerar den tid då sekvenseringsporen ändras från en adapter till en annan, (ii) en stall-signal som avser den tidsperiod då en DNA-sekvens befinner sig i porerna men ännu inte har rört sig, och (iii) signalspåren från DNA-sekvensering. En chimär läsning har däremot en stall-signal och en enorm spik-signal i mitten av läsningen. Chimeriska avläsningar kan ha två olika streckkodssekvenser i början och slutet, vilket gör det svårt att fastställa en streckkodskorg. Sammantaget visar dessa data på två kategorier av fel som bidrar till kontaminering av korsvisa prov i vårt dataset: (i) chimära läsningar (står för ∼80 % av alla korstilldelade läsningar) och (ii) läsningar med låg streckkodspoäng. För att förbättra kvaliteten på vårt slutliga dataset undersökte vi effekten av olika metoder för demultiplexering av streckkoder för att ta bort korstilldelade läsningar (tabell 3). Filtrering av de läsningar som har en intern adapter kan ta bort 90 % av de korsassocierade läsningarna och 24 % av de totala läsningarna. Vi testade också ett strängare filtreringsschema som krävde två streckkoder (en vardera i början och slutet av läsningen) för att göra en tilldelning. Detta tillvägagångssätt tog bort alla utom två korstilldelade läsningar, men förlorade 56 % av de totala läsningarna.

TABELL 3. Borttagning av korsassocierade läsningar och förlust av totala sekvenseringsdata genom två filtreringsmetoder med Porechop.
Vi undersöker också omfattningen av potentiella chimära läsningar i sekvenseringsdata. För enskilda sekvenseringskörningar av CHIKV, DENV och FLU-A visar kartläggningsresultaten att 2,3, 3,0 respektive 2,7 % av de kartlagda läsningarna har en kompletterande anpassning och anpassas minst två gånger till samma genom (tabell 4). Vi tar hänsyn till både streckkodsklassificerade och oklassificerade läsningar i multiplexsekvenseringsdata. Resultaten visar att 2,0 % av de kartlagda läsningarna har kompletterande anpassning och anpassades minst två gånger till samma genom, medan 0,052 % av de totala läsningarna anpassades till minst två olika genomer.

TABELL 4. Sammanfattning av antal och procentandel icke-kimeriska, självkimeriska och korskimeriska läsningar i varje sekvenseringskörning.
Diskussion
Det slutgiltiga målet för vår forskning är att utveckla en nanopore metagenomisk sekvenseringsbaserad diagnostisk test som möjliggör testning av infektionssjukdomar på plats i vården. Multiplexsekvensering ger möjlighet att förbättra skalbarheten och sänka kostnaderna, men korsprovkontaminering kan leda till fel i data och felaktig tolkning av resultaten.
I det här experimentet sammanförde vi rena bibliotek och utförde multiplex MinION-sekvensering för att undersöka omfattningen och källan till korsprovkontaminering. Vi identifierade att 0,056 % av de totala läsningarna korstilldelades till de felaktiga streckkodsbins, vilket är jämförbart med dem som rapporterats för Illumina-sekvenseringsplattformar från olika studier (mellan 0,06 och 0,25 %) (Nelson et al., 2014; D’Amore et al., 2016; Wright och Vetsigian, 2016). Våra resultat visade att chimära läsningar är den dominerande källan till fel i tilldelningen av korsvisa streckkoder. Korstilldelade chimära läsningar i det här datasetet kan bara ha bildats under sekvenseringen snarare än under biblioteksberedningen, eftersom de helt saknades i sekvenseringsdata för enskilda bibliotek, och det enda ytterligare bearbetningssteget var att blanda de slutliga sekvenseringsbiblioteken före laddning. Vi antar att den nuvarande algoritmen som implementerats i Albacore inte kan känna igen den korta dissociationen mellan DNA-sekvenser som löper samtidigt genom nanoporen och därmed sammanfoga mer än en sekvens i samma Fast5-fil.
Chimeriska läsningar observerades i MinION-sekvenseringsdata tidigare i White et al. (2017). Genom analyser av MinION-sekvenseringsdata från tre olika interferonamplikoner fann författarna att 1,7 % av de kartlagda läsningarna var chimärer. Våra resultat bidrar till den kunskap som stöder att chimärer är vanliga i MinION-sekvenseringsdata. Vi har identifierat att mellan 2 och 3 % av de totala läsningarna i tre individuella sekvenseringsdata och en multiplex-sekvenseringsdata är chimärer. Vår studie skiljer sig från tidigare arbete i följande två avseenden. För det första ger vi direkta bevis för att chimära reads kan bildas efter biblioteksberedning och under sekvensering; vi kopplade vidare dessa chimärer till kontaminering över provgränserna vid multiplex MinION-sekvensering enligt vad som diskuterats ovan. Å andra sidan har vår experimentuppsättning begränsningar när det gäller att identifiera potentiella chimärer som bildas under biblioteksförberedelsen, särskilt under adaptrelagering i standardprotokollet för multiplexsekvensering. För det andra återspeglar våra resultat den nuvarande statusen för MinION-sekvensering eftersom vi använde nyare och mest representativa ONT-sekvenseringskit, inklusive ligationssekvenseringskit 1D (SQK-LSK108) och native barcoding kit 1D (EXP-93 NBD103). Nanopore-sekvenseringstekniken är under snabb utveckling och förbättringar sker i alla avseenden. Nyare DNA-ligationssekvenseringskit (SQK-LSK109) och kit för direkt sekvensering av RNA (SQK-RNA001) har till exempel släppts. Algoritmen för basavrop som genomförts i Albacore och Guppy basecaller har uppgraderats. Alla dessa förändringar har effekt på omfattningen av chimärer i Nanopore-sekvenseringsdata och kontaminering av korsande streckkoder under multiplexsekvensering. Begränsningen i den här studien var det lilla antalet experiment, och ytterligare arbete med olika experimentupplägg skulle bidra till vår förståelse av Nanopore-multiplexsekvenseringsdata. Dessutom är det viktigt att undersöka bidragen från potentiella faktorer till kontaminering av tvärgående streckkoder, vilket skulle kasta ljus över bästa praxis för att analysera multiplexsekvenseringsdata.
Sammanfattningsvis visade vår studie att chimära läsningar är den dominerande källan till fel i tilldelningen av tvärgående streckkoder vid multiplex MinION-sekvensering. Den belyser behovet av noggrann filtrering av multiplex MinION-sekvenseringsdata före nedströmsanalys och den avvägning mellan känslighet och specificitet som gäller för streckkodsdemultiplexeringsmetoderna.
Författarbidrag
SP, KL, SL och YX utförde MinION-sekvensering. YX analyserade data. Alla författare utformade studien, deltog i tolkningen av resultaten och skrivandet av manuskriptet samt läste och godkände den slutliga versionen av detta manuskript.
Finansiering
Detta arbete stöddes av NIHR Oxford Biomedical Research Centre.
Intressekonfliktförklaring
Författarna förklarar att forskningen utfördes i avsaknad av kommersiella eller ekonomiska relationer som skulle kunna tolkas som en potentiell intressekonflikt.
Acknowledgments
Vi vill tacka Dr. Anthony Marriott (Public Health England) för att han tillhandahöll näsaspirat från illrar.
Supplementary Material
Footnotes
.