Introduction
Le syndrome de régression caudale (SRC) est un trouble peu fréquent décrit pour la première fois par Geoffroy Saint-Hilaire et Hohl en 1852, et en 1964 Duhmel a inventé le terme « syndrome de régression caudale ».1-3 Le SRC est un trouble causé par une anomalie des segments distaux de la colonne vertébrale, et il s’étend à un large éventail d’anomalies comme l’agénésie partielle de la moelle épinière, les malformations pelviennes associées, l’anus imperforé, les malformations génitales, les anomalies cardiaques, la dysplasie ou l’aplasie rénale bilatérale, l’hypoplasie pulmonaire, et la rotation externe extrême avec fusion des articulations inférieures, dont la forme la plus grave est la sirénomélie (syndrome de la sirène). Le SRC est également associé à une hypoplasie fémorale, des pieds déformés et une contracture de flexion des membres inférieurs. L’intelligence est préservée, en général.1,2,4 Il affecte entre 0,1 et 0,25 sur 10 000 grossesses, avec un rapport homme-femme de 2,7:1.3-5 Au niveau embryonnaire, on pense que le SRC est le résultat de défauts dans l’induction des éléments caudaux dans l’embryon avant le 28e jour de gestation. La lésion est produite dans l’axe mésodermique médian postérieur, entraînant l’absence de développement du vitellus mésoblastique caudal.4,6 L’étiologie exacte est inconnue ; cependant, le diabète maternel, la prédisposition génétique et l’hypoperfusion vasculaire ont été suggérés comme facteurs possibles4-7.
Le diabète prégestationnel est sans aucun doute un tératogène, et il existe de bonnes preuves que le diabète gestationnel peut être impliqué dans le développement de la forme la plus sévère du SRC.5
Pinter et reece ont prouvé que les altérations induites par l’hyperglycémie dans la fermeture du tube neural comprennent des cellules désordonnées, une diminution de la mitose et des changements qui indiquent une maturation prématurée. Le métabolisme oxydatif altéré par le diabète maternel peut provoquer une augmentation de la production de radicaux libres d’oxygène dans l’embryon en développement, ce qui peut être tératogène.8 Entre 16% et 22% des cas de SRC sont associés au diabète sucré maternel, ce qui augmente le risque d’avoir un enfant atteint de SRC jusqu’à 400.3,4,7 Plusieurs cas de familles atteintes de SRC ont été rapportés, ce qui suggère une possible transmission génétique avec différents modes de transmission possibles : Dominante liée au chromosome X, polygénique multifactorielle et autosomique dominante avec une pénétrance réduite et une expressivité variable.7 La théorie du » vol vasculaire » a été initialement proposée par Kampmeier en 1927 et réintroduite en 1986 par Stevenson. Adra et al, ont considéré la théorie du » vol vasculaire » de Stevenson comme une étiologie possible de la pathologie du SRC.7 Pendant le stade de développement embryonnaire, les structures plus caudales sont séparées des éléments céphaliques tels que le cerveau, la colonne vertébrale et la moelle épinière, d’où l’absence d’altérations cognitives dans ce syndrome.4
Il existe 2 groupes de SRC : le premier groupe est le plus touché avec la terminaison de la moelle épinière au-dessus de L1. Le sacrum se termine à S1, et dans certains cas, est absent. Les patients du second groupe présentent une dysgénésie moins sévère avec une implantation basse de la moelle épinière et attachée par un filum terminale épaissi ou un lipome intraspinal.4
Présentation du cas
Nous présentons le cas d’une fille nouveau-née d’une mère de 40 ans ayant des antécédents médicaux de fibromes utérins non traités, un passé de cycles gynécologiques irréguliers, 3 grossesses avec 2 accouchements et une cholécystectomie en 2008. Elle mentionne une grossesse non planifiée mais désirée, avec un suivi prénatal de grossesse à haut risque. Elle a assisté à plus de 15 consultations prénatales. À la 18e semaine de gestation, des données compatibles avec des malformations du tube neural avec agénésie sacrée ont été trouvées, ce qui a conduit à un diagnostic de SRC. Au cours du deuxième trimestre de la grossesse, une hyperglycémie a été détectée, elle a donc été placée sous traitement à l’insuline. À 40 semaines de gestation, une césarienne a été pratiquée pour obtenir un produit sain, avec un score d’Apgar de 8/9 et un âge gestationnel de 39,6 semaines. Lors de l’examen physique, nous avons observé un poids de 3,44 kg (7,58 lbs), une longueur de 49 cm (19,2 in), une circonférence de la tête de 35 cm (13,77 in) et une circonférence abdominale de 32 cm (12,59 in). Pendant l’inspection, nous avons pu observer une fœtopathie diabétique évidente, avec une pilosité abondante, et une implantation basse (Fig. 1). Fontanelle antérieure normale (2 x 2 cm), visage rond, pommettes saillantes, fissures palpébrales horizontales, arête nasale courte, bout rond, lèvres fines, palais plein, dysplasie des pavillons auriculaires avec hypertrichose de l’hélix, cou court avec un renflement dorsal, thorax normal, sans souffle à l’auscultation, abdomen souple sans masses palpables ni viscéromégalie, colonne vertébrale linéaire avec présence d’une fossette sur la peau au niveau de la région lombosacrée (Fig. 2), aucun os du sacrum n’était palpable, position normale de l’anus, perméable, avec un tonus absent. Il y avait un raccourcissement évident des membres inférieurs et un pied bot varus bilatéral (Fig. 3). Des radiographies ont été réalisées afin de voir les malformations osseuses, et nous avons pu observer une agénésie complète du sacrum avec une fusion lombo-iliaque. Une imagerie par résonance magnétique (IRM) du cerveau et de la colonne vertébrale (Fig. 4), a rapporté une corde abrupte et une terminaison lombaire au niveau L3. Après ce niveau, nous ne sommes en mesure d’identifier que des masses amorphes avec une intensité de signal graisseuse, liées au tissu sous-cutané. La moelle épinière est courte, le cône médullaire est plat au niveau T10, les deux os iliaques sont hypoplasiques et fusionnés au niveau moyen. Nous ne sommes pas en mesure de voir le sacrum. Il a été possible de voir les deux reins malrotés et hypoplasiques, une vessie urinaire proéminente compatible avec une vessie neurogène. L’image du cerveau a été rapportée comme normale. Une échographie de l’abdomen supérieur a été demandée afin d’exclure d’autres anomalies viscérales. Nous avons trouvé une discrète différence de taille du rein gauche, en plus de ne pas trouver sa configuration normale, ce qui suggère une discrète hypoplasie ainsi qu’une possible malrotation. Pendant son séjour, nous avons effectué une consultation multidisciplinaire, et des attelles plâtrées ont été placées sur les deux membres inférieurs. Le patient reste en suivi avec l’urologie, la traumatologie, la neurologie et la pédiatrie générale, ceci afin de surveiller de près tous les facteurs de risque et les éventuelles complications.
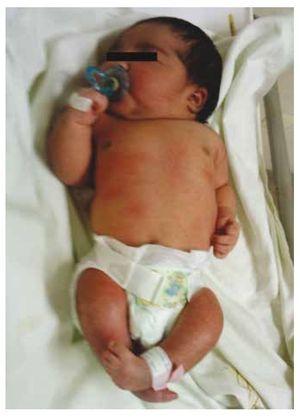
Figure 1 Nouveau-né présentant une fœtopathie diabétique.
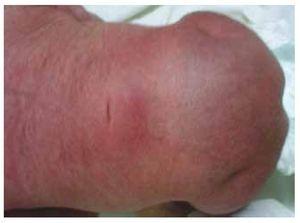
Figure 2 Une fossette sur la peau au niveau de la région lombosacrée.
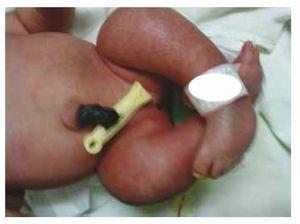
Figure 3 Equinovarus feet.
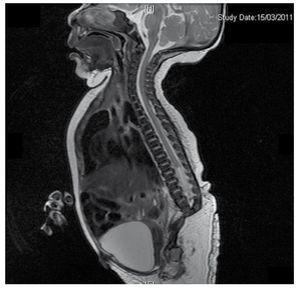
Figure 4 The lumbar spine magnetic resonance imaging (MRI) shows an abrupt chord and lumbar termination at the L3 level. Short spinal cord, flat conus medullaris at T10 level, hypoplastic iliac bones, fused at mid-level. No sacrum is seen.
Discussion
The CRS is a rare congenital malformation. Even though the specific etiologic factor is unknown, it is related to maternal diabetes, genetic predisposition and vascular hypoperfusion.3 Just as in the case presented, this alteration is characterized by agenesis of the sacrum involving iliac and lumbar vertebrae with their corresponding spinal segments and variable abnormalities in the lower limbs as well as in other organs. Fetal diagnosis tools allow early syndrome detection. L’échographie prénatale est l’instrument paraclinique le plus fréquemment utilisé ; un élément clé de l’échographie prénatale est l’évaluation détaillée de la colonne vertébrale et des membres inférieurs ; elle permet également de poser un diagnostic de SRC par la démonstration d’une terminaison abrupte de la colonne lombaire et de membres inférieurs hypoplasiques. Lors de l’établissement d’un diagnostic prénatal, nous devons nous attacher à discerner le degré de digénésie ainsi que les anomalies congénitales associées afin d’établir un pronostic et un plan opportun d’interventions thérapeutiques postnatales.3,5 La classification de Renshav, qui a été créée en 1978, classe le syndrome en 4 degrés en fonction de la gravité de l’agénésie du sacrum et de l’implication des vertèbres iliaques et lombaires.9 Selon cette classification, notre patient appartient au grade iV (agénésie complète du sacrum avec fusion des os iliaques). Ce groupe est associé à un pronostic encore plus défavorable avec un impact neurologique plus important et des séquelles multisystémiques, principalement au niveau rénal. Ce cas est un exemple clair du large éventail d’altérations qui peuvent affecter le fœtus en croissance à la suite d’un diabète maternel non contrôlé pendant la grossesse. En raison de la forte corrélation entre cette anomalie et la mère diabétique, et de son développement au cours des premiers stades de la grossesse, il est impératif de mettre en place une stratégie préventive comprenant un contrôle glycémique strict avant la période d’organogenèse embryonnaire, voire avant chez les patientes à haut risque. Une orientation adéquate et des examens génétiques prégestationnels sont également importants. Le traitement est un défi pour le médecin comme pour les parents, et il exige une approche multidisciplinaire impliquant un pédiatre, un chirurgien pédiatrique, un chirurgien orthopédique, un kinésithérapeute et un urologue en fonction de la gravité. Given the fact that the primary pathology is irreversible, treatment is just supportive, with the sole purpose of accomplishing a life as normal as possible.
Conflicts of interest
The authors have no conflicts of interest to declare.
Funding
No financial support was provided.
Received: September 2013;
Accepted: January 2014