Introduzione
La sindrome da regressione caudale (CRS) è un disturbo poco frequente descritto per la prima volta da Geoffroy Saint-Hilaire e Hohl nel 1852, e nel 1964 Duhmel ha coniato il termine “sindrome da regressione caudale”.1-3 La CRS è un disordine causato da un’anomalia dei segmenti spinali distali, e si estende ad una vasta gamma di anomalie come l’agenesia parziale del midollo spinale, malformazioni pelviche associate, ano imperforato, malformazioni genitali, anomalie cardiache, displasia o aplasia renale bilaterale, ipoplasia polmonare, ed estrema rotazione esterna con fusione delle articolazioni inferiori che sfocia nella forma più grave nella sirenomelia (sindrome della sirena). La CRS è anche associata a ipoplasia femorale, piedi deformi e contrattura da flessione degli arti inferiori. L’intelligenza è conservata, in generale.1,2,4 colpisce tra 0,1 e 0,25 su 10.000 gravidanze, con un rapporto maschio-femmina di 2,7:1.3-5 A livello embrionale, si ritiene che la CRS sia il risultato di difetti nell’induzione di elementi caudali nell’embrione prima del 28° giorno di gestazione. La lesione si produce nell’asse mesodermico mediale posteriore causando l’assenza di sviluppo del tuorlo mesoblastico caudale.4,6 L’esatta eziologia è sconosciuta; tuttavia, il diabete materno, la predisposizione genetica e l’ipoperfusione vascolare sono stati suggeriti come possibili fattori.4-7
Il diabete pre-gestazionale è senza dubbio un teratogeno, e ci sono buone prove che il diabete gestazionale può essere implicato nello sviluppo della forma più grave di CRS.5
Pinter e Reece hanno dimostrato che le alterazioni indotte dall’iperglicemia nella chiusura del tubo neurale includono cellule disordinate, diminuzione della mitosi, e cambiamenti che indicano una maturazione prematura. Il metabolismo ossidativo alterato dal diabete materno può causare un aumento della produzione di radicali senza ossigeno nell’embrione in via di sviluppo, che può essere teratogeno.8 Tra il 16% e il 22% dei casi di CRS sono associati al diabete mellito materno, che aumenta il rischio di avere un figlio con CRS fino al 400.3,4,7 Sono stati riportati diversi casi di famiglie con CRS, il che suggerisce una possibile trasmissione genetica con diverse modalità di trasmissione: X-linked dominante, poligenica multifattoriale, e modelli autosomici dominanti con penetranza ridotta ed espressività variabile.7 La teoria del “furto vascolare” fu inizialmente proposta da Kampmeier nel 1927 e reintrodotta nel 1986 da Stevenson. Adra et al. hanno considerato la teoria del “furto vascolare” di Stevenson come una possibile eziologia della patologia CRS.7 Durante la fase di sviluppo embrionale le strutture più caudali sono separate dagli elementi cefalici come il cervello, la spina dorsale e il midollo spinale, da qui la mancanza di alterazioni cognitive in questa sindrome.4
Ci sono 2 gruppi CRS: il primo gruppo è il più colpito con la terminazione del midollo spinale sopra L1. L’osso sacro termina a S1 e, in alcuni casi, è assente. I pazienti del secondo gruppo mostrano una disgenesi meno grave con un basso impianto del midollo spinale e legato da un filum terminale ispessito o da un lipoma intraspinale.4
Presentazione del caso
Presentiamo il caso di una neonata da una madre di 40 anni con una storia medica di fibromi uterini non trattati, uno sfondo di cicli ginecologici irregolari, 3 gravidanze con 2 parti e colecistectomia nel 2008. Parla di una gravidanza non pianificata ma voluta, con un’assistenza prenatale di gravidanza ad alto rischio. Ha partecipato a più di 15 consulti prenatali. Alla 18° settimana di gestazione sono stati trovati dati compatibili con malformazioni del tubo neurale con agenesia sacrale, con conseguente diagnosi di CRS. Durante il secondo trimestre di gravidanza è stata rilevata un’iperglicemia, quindi è stata messa in trattamento con insulina. A 40 settimane di gestazione è stato eseguito un taglio cesareo ottenendo un prodotto sano, con un punteggio Apgar di 8/9 con un’età gestazionale di 39,6 settimane. All’esame fisico abbiamo osservato un peso di 3,44 kg (7,58 lbs), una lunghezza di 49 cm (19,2 in), una circonferenza della testa di 35 cm (13,77 in) e una circonferenza addominale di 32 cm (12,59 in). Durante l’ispezione abbiamo potuto osservare un’evidente fetopatia diabetica, con abbondanti peli, e basso impianto (Fig. 1). Fontanella anteriore normale (2 x 2 cm), viso rotondo, zigomi prominenti, fessure palpebrali orizzontali, ponte nasale corto, punta rotonda, labbra sottili, palato pieno, displasia dei padiglioni auricolari con ipertricosi dell’elice, collo corto con un rigonfiamento dorsale, torace normale, senza mormorii durante l’auscultazione, l’addome era morbido senza masse palpabili o visceromegalia, spina dorsale lineare con presenza di una fossetta sulla pelle nella regione lombosacrale (Fig. 2), nessun osso sacro era palpabile, posizione normale dell’ano, permeabile, con un tono assente. C’era un evidente accorciamento degli arti inferiori e piede torto bilaterale (Fig. 3). Sono state fatte delle radiografie per vedere le malformazioni ossee, e abbiamo potuto osservare un’agenesia completa dell’osso sacro con una fusione lombare-iliaca. Una risonanza magnetica (RMN) del cervello e della colonna vertebrale (Fig. 4), ha segnalato una brusca corda e una terminazione lombare a livello di L3. Dopo questo livello siamo in grado di identificare solo masse amorfe con intensità di segnale grasso, relative al tessuto sottocutaneo. Midollo spinale corto, cono midollare piatto a livello di T10, entrambe le ossa iliache erano ipoplasiche e fuse a livello medio. Non siamo in grado di vedere l’osso sacro. È stato possibile vedere entrambi i reni malrotati e ipoplasici, una vescica urinaria prominente compatibile con una vescica neurogena. L’immagine cerebrale è stata riportata come normale. Fu richiesta un’ecografia dell’addome superiore per escludere altre anomalie viscerali trovando una discreta discrepanza nelle dimensioni del rene sinistro, oltre a non trovare la sua normale configurazione, il che suggerì una discreta ipoplasia oltre che una possibile malrotazione. Durante la degenza abbiamo effettuato un consulto multidisciplinare e sono state messe stecche di gesso su entrambi gli arti inferiori. Il paziente rimane in follow-up con Urologia, Traumatologia, Neurologia e Pediatria generale, questo per tenere sotto controllo tutti i fattori di rischio e le possibili complicazioni.
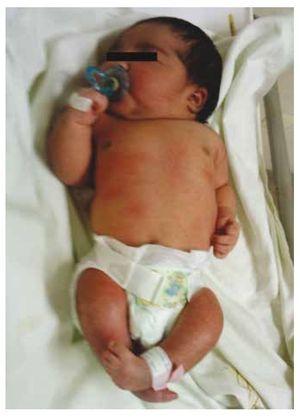
Figura 1 Neonato con fetopatia diabetica.
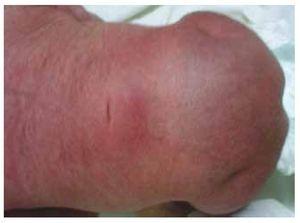
Figura 2 Una fossetta sulla pelle nella regione lombosacrale.
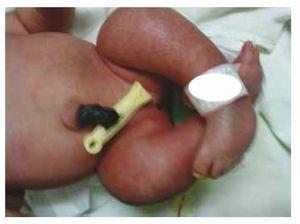
Figure 3 Equinovarus feet.
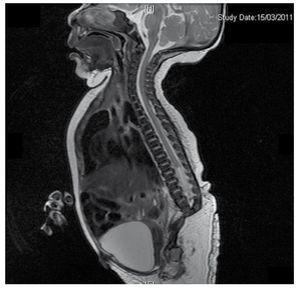
Figure 4 The lumbar spine magnetic resonance imaging (MRI) shows an abrupt chord and lumbar termination at the L3 level. Short spinal cord, flat conus medullaris at T10 level, hypoplastic iliac bones, fused at mid-level. No sacrum is seen.
Discussion
The CRS is a rare congenital malformation. Even though the specific etiologic factor is unknown, it is related to maternal diabetes, genetic predisposition and vascular hypoperfusion.3 Just as in the case presented, this alteration is characterized by agenesis of the sacrum involving iliac and lumbar vertebrae with their corresponding spinal segments and variable abnormalities in the lower limbs as well as in other organs. Fetal diagnosis tools allow early syndrome detection. L’ecografia prenatale è lo strumento paraclinico più utilizzato; un elemento chiave dell’ecografia prenatale è la valutazione dettagliata della colonna vertebrale e degli arti inferiori; permette anche una diagnosi di CRS attraverso la dimostrazione di una brusca terminazione della colonna lombare e di arti inferiori ipoplasici. Quando si fa una diagnosi prenatale, dobbiamo concentrarci sul discernimento del grado di digenesi così come delle anomalie congenite associate allo scopo di stabilire una prognosi e un piano tempestivo di interventi terapeutici postnatali.3,5 La classificazione di Renshav, creata nel 1978, categorizza la sindrome in 4 gradi in base alla gravità dell’agenesia del sacro e al coinvolgimento delle vertebre iliache e lombari.9 Secondo questa classificazione, il nostro paziente appartiene al grado iV (agenesia completa del sacro con fusione delle ossa iliache). Questo gruppo è associato a una prognosi ancora peggiore con un maggiore impatto neurologico e sequele multisistemiche, soprattutto a livello renale. Questo caso è un chiaro esempio della vasta gamma di alterazioni che possono colpire il feto in crescita come conseguenza del diabete materno incontrollato durante la gravidanza. A causa dell’alta correlazione tra questo difetto e la madre diabetica, e il suo sviluppo durante le prime fasi della gravidanza, è imperativo avere una strategia preventiva che includa uno stretto controllo glicemico prima del periodo dell’organogenesi embrionale, o anche prima in pazienti ad alto rischio. Anche una guida adeguata e gli esami genetici pre-gestazionali sono importanti. Il trattamento è una sfida sia per il medico che per i genitori, e richiede un approccio multidisciplinare che coinvolge un pediatra, un chirurgo pediatrico, un ortopedico, un fisioterapista e un urologo a seconda della gravità. Given the fact that the primary pathology is irreversible, treatment is just supportive, with the sole purpose of accomplishing a life as normal as possible.
Conflicts of interest
The authors have no conflicts of interest to declare.
Funding
No financial support was provided.
Received: September 2013;
Accepted: January 2014