Rapport de cas – Recherche biomédicale (2019) Volume 30, numéro 2
Rapport d’un cas mortel de syndrome de potter : Un rapport de cas.
Mehrbano Amirshahi, Mahin Badakhsh* et Zohreh Sadat Hashemi
Département des sages-femmes, École d’infirmières et de sages-femmes, Université des sciences médicales de Zabol, Zabol, Iran
*Auteur correspondant : Mahin Badakhsh
Département de sages-femmes
École d’infirmières et de sages-femmes
Université des sciences médicales de Zabol
Iran
Date d’acceptation : 21 février 2019
DOI : 10.35841/biomedicalresearch.30-19-084
Visitez pour plus d’articles connexes à Biomedical Research
Abstract
Contexte : Le syndrome de Potter est un trouble congénital rare qui désigne un ensemble de manifestations cliniques associées à un oligohydramnios causé par une insuffisance rénale fœtale. La caractéristique de ce syndrome est le tableau clinique particulier qui, en plus de l’oligohydramnios, présente une hypoplasie pulmonaire, une agénésie rénale bilatérale, des déformations des membres et un visage spécifique. L’embryon meurt avant ou immédiatement après la naissance en raison d’une insuffisance respiratoire. Le but de cette étude est de rapporter un fœtus avec le syndrome de Potter qui est né par accouchement vaginal. Rapport de cas : L’examen échographique de la femme enceinte a révélé que son fœtus de sexe masculin, dont l’âge gestationnel était de 25 semaines, présentait le syndrome de Potter et que l’indice du liquide amniotique était nul. La mère a été hospitalisée en unité de travail et a accouché par voie naturelle. Le bébé présentait un tableau clinique de syndrome de Potter et une détresse respiratoire sévère et est décédé peu après la naissance. Conclusion : Le syndrome de Potter est une affection très grave et la plupart du temps mortelle. L’échographie prénatale en examinant l’oligohydramnios et les reins aide au diagnostic.
Mots clés
Syndrome de Potter, Oligohydramnios, Hypoplasie pulmonaire, Fœtus
Introduction
Le syndrome de Potter est une malformation congénitale rare qui touche principalement les fœtus mâles et se caractérise par une hypoplasie pulmonaire causée par une insuffisance rénale. Il a été signalé pour la première fois par Edith Potter en 1946. Après la 16e semaine de grossesse, la quantité de liquide amniotique dépend principalement de la production d’urine par le fœtus. Pendant la vie intra-utérine, le fœtus avale continuellement le liquide amniotique qui est renvoyé dans le sac amniotique par les reins. L’oligohydramnios se produit lorsque le volume du liquide amniotique est inférieur à la normale pour cette période de la grossesse. Cette diminution du volume peut être due à une réduction du débit urinaire, secondaire à des causes telles qu’une agénésie rénale bilatérale, une obstruction des voies urinaires et une rupture prolongée des sacs amniotiques. L’urine du fœtus, essentielle au développement des poumons, joue son rôle en contribuant au développement des voies respiratoires et des alvéoles, en créant une pression hydraulique et en fournissant de la proline, un acide aminé essentiel au développement des poumons. Si les alvéoles et, par conséquent, les poumons ne sont pas suffisamment développés à la naissance, le nouveau-né ne pourra pas bien respirer et souffrira d’une détresse respiratoire due à une hypoplasie pulmonaire. Par conséquent, secondaire à l’insuffisance rénale, l’hypoplasie pulmonaire est la principale cause de décès chez les nouveau-nés atteints du syndrome de Potter .
La production d’urine par l’embryon affecte non seulement le volume du liquide amniotique, mais préserve également l’embryon contre la pression de la paroi utérine de la mère comme un coussin. L’oligohydramnios entraîne une forme particulière de fœtus appelée « faciès de Potter », qui se caractérise par des traits tels qu’une arête nasale aplatie, un menton hypoplasique, des plis épicanthaux, une cataracte et des oreilles basses .
La cause principale du syndrome de Potter est inconnue ; ce syndrome a un fond génétique dans certains cas, et est plus fréquent chez les nouveau-nés ayant des antécédents familiaux d’anomalies rénales . Le syndrome, dont l’incidence est de 1 pour 2 000 à 5 000 fœtus, est associé à un risque de récurrence de 3 à 6 %, et est retrouvé dans 0,2 à 0,4 % des autopsies chez les nouveau-nés décédés ou ceux qui meurent immédiatement après la naissance . Cependant, on pense que la maladie peut avoir une prévalence plus élevée parce que les fœtus affectés sont nés morts ou meurent peu après la naissance. Il n’existe aucune méthode connue pour prévenir cette maladie mortelle. Par conséquent, une échographie de dépistage est recommandée à 16-18 semaines de gestation pour les mères ayant des antécédents positifs de grossesse pour ce syndrome afin d’évaluer l’oligohydramnios et les reins du fœtus. Bien que ce syndrome ait des conséquences mortelles, qu’il ne soit pas compatible avec la vie et qu’il se termine par la mort du bébé, les fœtus affectés nécessitent une réanimation et un traitement de l’obstruction du débit urinaire au moment de l’accouchement. La présente étude visait à rapporter un fœtus atteint du syndrome de Potter qui est né par un accouchement normal.
Rapport de cas
Le cas était un fœtus masculin de 25 semaines atteint du syndrome de Potter, né par un accouchement normal. La mère a été admise à la maternité de l’hôpital Amir al-Momenin, Zabol, Iran, le 10 mai 2017 avec une échographie, qui a montré l’absence de liquide amniotique et le diagnostic de syndrome de Potter pour le fœtus. La mère était âgée de 24 ans, iranienne, femme au foyer, gravide 3 et para 1, et avait des antécédents d’avortement au premier mois. Elle ne se souvenait pas du premier jour de ses dernières menstruations, et son âge gestationnel était de 25 semaines selon la seule échographie réalisée le même jour. Ses antécédents d’accouchement étaient normaux (son enfant précédent était en bonne santé et a eu un accouchement normal), et elle avait reçu des soins prénataux pendant sa grossesse. Ses tests de grossesse étaient normaux. Il n’y avait pas d’antécédents de tabagisme, de drogues ou d’alcool, de maladies sous-jacentes telles que le diabète, l’hypertension, les maladies thyroïdiennes, cardio-pulmonaires et rénales, d’infections, et de consommation de médicaments autres que les médicaments de grossesse habituels (suppléments de fer et de vitamines). La mère et son mari étaient cousins et elle n’a pas mentionné d’antécédents familiaux d’une naissance néonatale avec un trouble similaire. Les tests sérologiques prénataux étaient négatifs pour la syphilis, le SIDA, l’hépatite B et la rubéole. Elle avait un groupe sanguin B positif.
Selon l’échographie mentionnée, l’âge gestationnel était de 25 semaines, l’indice du liquide amniotique était nul, les reins du fœtus étaient invisibles dans la position anatomique normale, la vessie était invisible, et il y avait une restriction de croissance fœtale sévère et une cardiomégalie sévère (plus de 80%). The diagnosis was Potter syndrome based on ultrasound, after which the mother was taken to the maternity ward.
The mother was hospitalized and, after the normal course of labor, gave birth to a male new-born through vaginal method. The boy had growth limitation (800 g birth weight), the clinical presentation of Potter syndrome, and severe respiratory distress and died immediately after delivery (Figures 1 and 2).
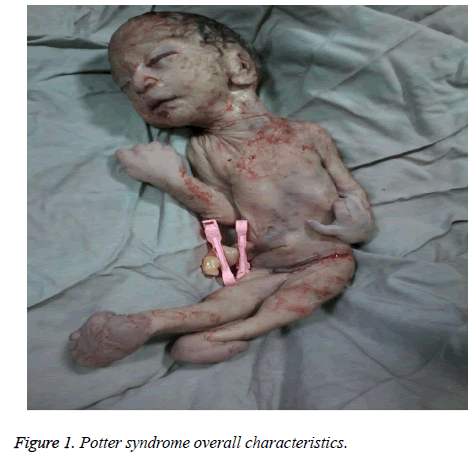
Figure 1: Potter syndrome overall characteristics.
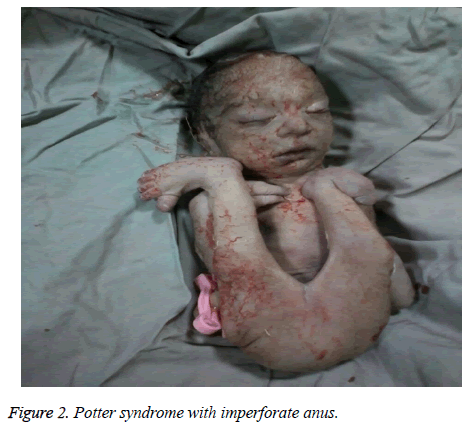
Figure 2: Potter syndrome with imperforate anus.
Discussion
The present study introduced a rare case that has not been reported in the region so far. It was a fatal case of Potter syndrome that was born through normal delivery. L’échographie de la femme a révélé un fœtus à 25 semaines de grossesse avec un syndrome de Potter et un indice de liquide amniotique de zéro. La mère a été admise à l’unité d’accouchement et le bébé est né par voie vaginale. Le nouveau-né présentant la présentation clinique du syndrome de Potter présentait une détresse respiratoire sévère et est décédé peu après la naissance.
Le syndrome de Potter, décrit pour la première fois par Edith Potter chez des nouveau-nés, se caractérise par une agénésie rénale bilatérale ou d’autres anomalies rénales telles qu’une aplasie, une dysplasie, une hypoplasie ou une polykystose rénale. Il est reconnu davantage chez les garçons, avec un rapport homme/femme de 2 à 1, ce qui suggère la présence de certains gènes sur le chromosome Y . Le syndrome de Potter peut être observé chez des nouveau-nés avec des reins normaux, mais la mère avait une fuite de liquide amniotique chronique et prolongée pendant les semaines intermédiaires de la grossesse .
Dans cette étude, l’échographie des femmes enceintes a révélé un cas de syndrome de Potter chez un fœtus mâle à la 25e semaine de grossesse, un indice de liquide amniotique nul et l’absence des deux reins et de la vessie. Les résultats de l’échographie de fœtus atteints d’une grave maladie rénale dans 23 familles indiquaient un oligohydramnios prolongé, un dysfonctionnement rénal grave et un faciès de Potter, et les nourrissons sont morts quelques heures ou quelques jours après la naissance. Moreira et Reuvers ont également souligné la naissance de fœtus atteints du syndrome de Potter .
Les caractéristiques néonatales du syndrome de Potter comprennent des altérations faciales, des malformations des membres, une limitation de la croissance fœtale et une hypoplasie pulmonaire, connues sous le nom de tétrade d’oligohydramnios. Ces caractéristiques résultent d’une compression fœtale due à un oligohydramnios prolongé. Le faciès de Potter se caractérise par un hypertélorisme, une paupière mongole, un pli épicanthique, une arête nasale et un lobe d’oreille aplatis, un nez en bec de perroquet, des oreilles basses, un menton en retrait, un petit pli sous la lèvre, un cou court et des chaînes supplémentaires autour du cou. La présentation clinique du nourrisson de cette étude était la suivante : visage en retrait, arête nasale et lobe de l’oreille aplatis, nez en bec de perroquet, oreilles décollées, menton petit, court et en retrait, petit pli sous la lèvre, cou court et plis supplémentaires autour du cou, mains larges, déviation des mains au niveau du poignet, valgus bilatéral et limitation sévère de la croissance fœtale, ce qui a entraîné un poids de 800 g à la naissance. Ces manifestations cliniques étaient conformes à celles de Shastry et Al-Haggar .
La pression constante des parois utérines sur la paroi thoracique du fœtus et la pression des organes intra-abdominaux sur le diaphragme constituent l’une des conséquences de l’oligohydramnios dans le syndrome de Potter, qui sont également les principales causes de l’hypoplasie et de l’insuffisance pulmonaire dans ce syndrome. La gravité de l’hypoplasie pulmonaire dépend de la phase de développement du poumon dans laquelle se produit l’oligohydramnios, ainsi que de l’intensité et de la durée de l’oligohydramnios. En raison de la détresse respiratoire grave et de l’hypoplasie pulmonaire, les fœtus naissent avec des reins dysplasiques ou meurent peu après la naissance . Dans cette étude, l’indice du liquide amniotique était nul selon l’échographie et le nouveau-né a présenté une détresse respiratoire sévère à la naissance et est décédé quelques instants après la naissance.
Le syndrome de Potter peut être associé à des anomalies cardiaques congénitales, des anomalies gastro-intestinales (telles que l’atrésie de l’œsophage, l’agénésie colique et les malformations anales et duodénales, le diverticule de Meckel et les kystes du pancréas et de la rate), des troubles squelettiques, des anomalies cérébrales et diverses associations, comme Vakarl et al . Dans la présente étude, l’échographie a signalé une cardiomégalie sévère (plus de 80 %) et une vessie invisible ; et l’examen physique du nouveau-né a révélé des anomalies des membres et un anus imperforé.
La cause principale du syndrome de Potter reste incertaine dans la plupart des cas, mais il a une raison génétique dans certains cas, et le schéma d’héritage dépend d’une cause génétique particulière. Les anomalies génétiques telles que l’hérédité autosomique dominante ou récessive de la polykystose rénale, la dysplasie rénale héréditaire, causée par des mutations des gènes RET et UPK3A et des anomalies chromosomiques, peuvent entraîner des anomalies de développement et conduire au syndrome de Potter. Ce syndrome se produit sporadiquement mais peut être hérité lorsqu’il est issu de la triade autosomique dominante. Le syndrome de Potter est plus fréquent chez les nourrissons qui ont des antécédents familiaux d’anomalies rénales. Dans la présente étude, aucune cause spécifique n’a été trouvée pour cette maladie, et il n’y avait pas d’antécédents familiaux d’anomalies rénales. San et Samal ont également souligné l’étiologie inconnue du syndrome de Potter et ses raisons génétiques dans leurs études .
Bien que la principale cause du syndrome de Potter soit l’anomalie du développement des reins, le diagnostic initial est effectué par une échographie qui peut montrer la perte ou l’absence de liquide amniotique et l’absence de vessie, et se poursuit par une investigation sur la présence ou l’absence de reins. Le conseil génétique est important pour confirmer le diagnostic. L’identification de ce syndrome, qui est caractérisé par une agénésie rénale bilatérale, une hypoplasie pulmonaire, un faciès de potter et une malformation des membres, peut être problématique par l’échographie en raison de l’oligohydramnios . Dans cette étude, l’échographie a montré l’absence d’indice de liquide amniotique, l’invisibilité de la vessie et l’agénésie rénale bilatérale.
Le syndrome de Potter est incompatible avec la vie et a un pronostic mortel. La grossesse peut être interrompue avant que le fœtus n’atteigne le stade de la vie. Les soins standard de la grossesse ne changent pas lorsqu’il est décidé de poursuivre la grossesse . L’une des limites de la présente étude est que le test génétique n’a pas été effectué, car les parents n’ont pas accepté l’autopsie.
Dans la présente étude, une mère dans sa 25e semaine de grossesse avec un fœtus atteint du syndrome de Potter, diagnostiqué par échographie, a été admise à l’unité d’accouchement et a donné une naissance normale à un bébé qui est décédé quelques instants après la naissance.
Conclusion
Le syndrome de Potter est une maladie très grave et une condition mortelle. L’échographie prénatale permet de détecter ce syndrome en examinant l’oligohydramnios et les affections rénales.
Conflit d’intérêt
Les auteurs déclarent n’avoir aucun conflit d’intérêt concernant la publication de ce rapport de cas.
Financement
Aucun financement n’a été recherché ou obtenu en relation avec ce rapport de cas.
Consentement du patient
Obtenu
Providence et examen par les pairs
Ce rapport de cas a été examiné par les pairs.
- Potter EL. Caractéristiques faciales des nourrissons présentant une agénésie rénale bilatérale. Am J Obstet Gynecol 1946 ; 51 : 885-888.
- Srihari Babu M, Asha Latha D, Radhika P. Potters syndrome : Un rapport de cas. JDMS 2015 ; 14 : 14-16.
- Soudhanshu Kumar D, Sidharth Sankar M. Faciès de Potter avec maladie rénale polykystique en association avec d’autres anomalies congénitales : deux rapports de cas. J Med Dent Sci 2013 ; 2 : 2665-2668.
- Khatami F. Le syndrome de Potters : Une étude de 15 patients. Arch Iranian Med 2004 ; 7 : 186- 189.
- Sarkar S, Das Gupta S, Barua M, Ghosh R, Mondal K, Chatterjee U, Datta C. La séquence de Potters : Une histoire du rare, du plus rare et du plus rare. Indian J Pathol Microbiol 2015 ; 58 : 102-104.
- Manoj MG, Kakkar S. Le syndrome de Potters – une constellation fatale d’anomalies. Indian J Med Res 2014 ; 139 : 648-649.
- Himabindu A, Narasinga B. Un cas fatal de syndrome de potters-un rapport de cas. JCDR 2011 ; 5 : 1264-1266.
- Moreira BL, Monarim MA, Romano RF, Mattos LA, DIppolito G. Doege-Potter syndrome. Radiol Bras 2015 ; 48 : 195-196.
- Reuvers JR, van Dorp M, Van Schil PE. Tumeur fibreuse solitaire de la plèvre avec syndrome de Doege-Potter associé. Acta Chir Belg 2016 ; 116 : 386-387.
- Shastry SM, Kolte SS, Sanagapati PR. La séquence de Potters. J Clin Neonatol 2012 ; 1 : 157-159.
- Al-Haggar M, Yahia S, Abdel-Hadi D, Grill F, Al-Kaissi A. Sirénomélie (symélie apus) avec syndrome de Potters en relation avec un diabète sucré gestationnel : Un rapport de cas et une revue de la littérature. Afr Health Sci 2010 ; 10 : 395-399.
- Dhundiraj KM, Madhukar DN, Ambadasrao PG, Wamanrao KS, Prem ZM. Le syndrome de Potters : Un rapport de 5 cas. Indian J Pathol Microbiol 2006 ; 49 : 254-257.
- Dayal J, Maheshwari E, Ghai PS, Bhatotia S. Antenatal ultrasound diagnosis of Potters syndrome. Indian J Radiol Imaging 2003 ; 13 : 81-83.
- San Agustin JT, Klena N, Granath K, Panigrahy A, Stewart E, Devine W, Strittmatter L, Jonassen JA, Liu X. Lien génétique entre les anomalies congénitales rénales et les cardiopathies congénitales. Nat Commun 2016 ; 22 : 111-118.
- Samal SK, Rathod S. Sirenomelia : le syndrome de la sirène : rapport de deux cas. J Nat Sci Biol Med 2015 ; 6 : 264-266.
- Gerards FA, Twisk JW, Fetter WP. Prédire l’hypoplasie pulmonaire avec une échographie en 2 ou 3 dimensions dans les grossesses compliquées. Am J Obstet Gynecol 2008; 198: 140-146.
- Kemper MJ, Mueller-Wiefel DE. Prognosis of antenatally diagnosed oligohydramnios of renal origin. Eur J Pediatr 2007; 166: 393-398.