Raport de caz – Cercetări biomedicale (2019) Volumul 30, Numărul 2
Raportul unui caz mortal de sindrom potter: Un raport de caz.
Mehrbano Amirshahi, Mahin Badakhsh* și Zohreh Sadat Hashemi
Departamentul de moașe, Școala de asistență medicală și moașe, Universitatea de științe medicale Zabol, Zabol, Iran
*Autor corespondent: Mahin Badakhsh
Departamentul de moașe
Școala de asistență medicală și moașe
Universitatea de științe medicale Zabol
Iran
Data acceptării: February 21, 2019
DOI: 10.35841/biomedicalresearch.30-19-084
Vizitați pentru mai multe articole conexe la Biomedical Research
Abstract
Background: Sindromul Potter este o afecțiune congenitală rară care se referă la un set de manifestări clinice care sunt asociate cu oligohidramniosul cauzat de insuficiența renală fetală. Semnul distinctiv al acestui sindrom este tabloul clinic special care pe lângă oligohidramniosul caracteristic cu hipoplazie pulmonară, agenezie renală bilaterală; deformări ale membrelor și față specifică. Embrionul moare înainte sau imediat după naștere din cauza insuficienței respiratorii. Scopul acestui studiu este de a raporta un făt cu sindromul Potter care s-a născut prin naștere vaginală. Raport de caz: Examinarea ecografică a femeii însărcinate a arătat că fătul de sex masculin cu vârsta gestațională de 25 de săptămâni cu sindromul Potter, iar indicele de lichid amniotic este zero. Mama a fost spitalizată în travaliu unitar și a născut pe cale naturală. Bebelușul a prezentat un tablou clinic de sindrom Potter și detresă respiratorie severă și a decedat la scurt timp după naștere. Concluzii: Sindromul Potter este o afecțiune foarte gravă și de cele mai multe ori este mortală. Ecografia prenatală prin examinarea oligohidramniosului și a rinichilor ajută la diagnosticare.
Cuvintele cheie
Sindromul Potter, Oligohidramnios, Hipoplazie pulmonară, Fetus
Introducere
Sindromul Potter este o malformație congenitală rară care afectează în principal fetușii de sex masculin și se caracterizează prin hipoplazie pulmonară cauzată de insuficiență renală. A fost raportat pentru prima dată de Edith Potter în 1946. După a 16-a săptămână de sarcină, cantitatea de lichid amniotic depinde în principal de producția de urină de către făt. În timpul vieții intrauterine, fătul înghite continuu lichidul amniotic, care se întoarce înapoi în sacul amniotic prin rinichi. Oligohidramniosul apare atunci când volumul de lichid amniotic este mai mic decât cel normal pentru acea perioadă a sarcinii. Această scădere a volumului se poate datora unei reduceri a debitului urinar, secundară unor cauze precum agenezia renală bilaterală, obstrucția tractului urinar și ruptura prelungită a sacilor amniotici. Urina fătului, esențială pentru dezvoltarea plămânilor, își joacă rolul de a contribui la dezvoltarea căilor respiratorii și a alveolelor, de a crea presiune hidraulică și de a furniza prolină, un aminoacid esențial pentru dezvoltarea plămânilor. Dacă alveolele și, ca urmare, plămânii nu sunt dezvoltate în mod adecvat la naștere, nou-născutul nu va putea respira bine și va suferi de detresă respiratorie din cauza hipoplaziei pulmonare. Prin urmare, secundar insuficienței renale, hipoplazia pulmonară este principala cauză de deces la nou-născuții cu sindromul Potter .
Producția de urină de către embrion nu numai că afectează volumul lichidului amniotic, dar, de asemenea, conservă embrionul împotriva presiunii peretelui uterin al mamei, ca o pernă. Oligohidramniosul are ca rezultat o formă specială de făt numită „facies Potter”, care se caracterizează prin trăsături precum punte nazală aplatizată, bărbie hipoplastică, pliuri epicantale, cataractă și urechi joase .
Cauza principală a sindromului Potter este necunoscută; acest sindrom are un fond genetic în unele cazuri și este mai frecvent la nou-născuții cu antecedente familiale de anomalii renale . Sindromul, cu o incidență de 1 la fiecare 2.000 până la 5.000 de fetuși, este asociat cu un risc de recurență de 3-6%, și se găsește în 0,2-0,4% din autopsii la nou-născuții morți sau la cei care mor imediat după naștere . Cu toate acestea, se crede că boala poate avea o prevalență mai mare deoarece fetușii afectați se nasc morți sau mor la scurt timp după naștere. Nu se cunoaște nicio metodă de prevenire a acestei boli mortale. Prin urmare, se recomandă ecografia de screening la 16-18 săptămâni de gestație pentru mamele cu antecedente pozitive de sarcină pentru acest sindrom, cu scopul de a evalua oligohidramniosul și rinichii fetali. Deși acest sindrom are consecințe mortale și nu este compatibil cu viața și se termină cu moartea copilului, fetușii afectați necesită resuscitare și tratamentul obstrucției de ieșire urinară în momentul nașterii . Studiul de față și-a propus să raporteze un făt cu sindrom Potter care s-a născut prin naștere normală.
Raport de caz
Cazul a fost un făt de sex masculin în vârstă de 25 de săptămâni cu sindrom Potter, născut prin naștere normală. Mama a fost internată la maternitatea Spitalului Amir al-Momenin din Zabol, Iran, la 10 mai 2017, cu o ecografie, care a arătat absența lichidului amniotic și diagnosticul de sindrom Potter pentru făt. Mama era în vârstă de 24 de ani, iraniană, casnică, gravidă 3 și para 1 și avea antecedente de avort în luna 1. Nu-și amintea prima zi a ultimei menstruații, iar vârsta gestațională era de 25 de săptămâni, conform singurei ecografii efectuate în aceeași zi. Avea un istoric de naștere normal (copilul ei anterior a fost sănătos și a avut o naștere normală) și primise îngrijire prenatală în timpul sarcinii. Testele ei de sarcină au fost normale. Nu existau antecedente de fumat, droguri sau alcool, boli de bază, cum ar fi diabetul, hipertensiunea, tiroida, cardiopulmonare și renale, infecții și utilizarea de medicamente în afară de medicamentele de rutină pentru sarcină (suplimente de fier și vitamine). Mama și soțul ei erau verișori și nu a menționat un istoric familial al unei nașteri neonatale cu o afecțiune similară. Testele serologice prenatale au fost negative pentru sifilis, SIDA, hepatită B și rubeolă. Avea grupa sanguină B pozitiv.
Conform ecografiei menționate, vârsta gestațională era de 25 de săptămâni, indicele de lichid amniotic era zero, rinichii fătului erau invizibili în poziție anatomică normală, vezica urinară era invizibilă și existau o restricție severă de creștere fetală și o cardiomegalie severă (peste 80%). The diagnosis was Potter syndrome based on ultrasound, after which the mother was taken to the maternity ward.
The mother was hospitalized and, after the normal course of labor, gave birth to a male new-born through vaginal method. The boy had growth limitation (800 g birth weight), the clinical presentation of Potter syndrome, and severe respiratory distress and died immediately after delivery (Figures 1 and 2).
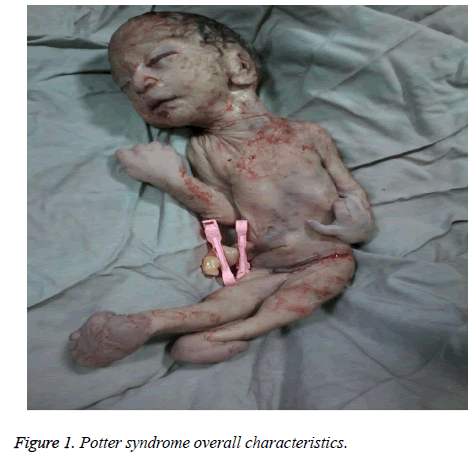
Figure 1: Potter syndrome overall characteristics.
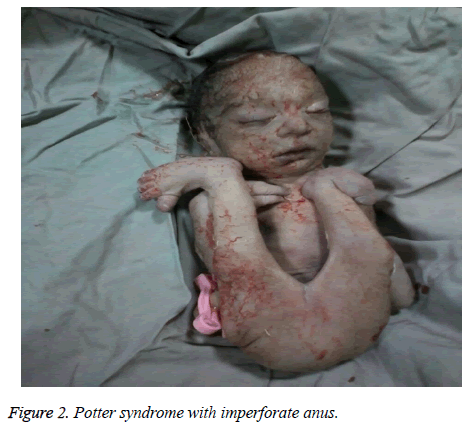
Figure 2: Potter syndrome with imperforate anus.
Discussion
The present study introduced a rare case that has not been reported in the region so far. It was a fatal case of Potter syndrome that was born through normal delivery. Ecografia femeii a evidențiat un făt la 25 de săptămâni de sarcină cu sindrom Potter și un indice de lichid amniotic de zero. Mama a fost internată în unitatea de naștere, iar copilul s-a născut pe cale vaginală. Nou-născutul cu prezentarea clinică a sindromului Potter a avut detresă respiratorie severă și a murit la scurt timp după naștere.
Sindromul Potter, descris pentru prima dată de Edith Potter la nou-născuți, se caracterizează prin agenezie renală bilaterală sau alte anomalii renale, cum ar fi aplasia, displazia, hipoplazia sau boala polichistică renală. Este recunoscut mai mult la băieți, cu un raport masculin/feminin de 2 la 1, sugerând prezența anumitor gene pe cromozomul Y . Sindromul Potter poate fi observat la nou-născuții cu rinichi normali, dar mama a avut o scurgere cronică și prelungită de lichid amniotic în timpul săptămânilor mijlocii de sarcină .
În acest studiu, ecografia femeilor însărcinate a relevat un caz de sindrom Potter la un făt de sex masculin în săptămâna 25 de sarcină, un indice de lichid amniotic zero și lipsa ambilor rinichi și a vezicii urinare. Constatările ecografice ale fetușilor cu afecțiuni renale severe din 23 de familii au indicat oligohidramnios prelungit, disfuncție renală severă și facies Potter, iar copiii au murit la câteva ore sau zile după naștere. Moreira și Reuvers au evidențiat, de asemenea, nașterea unor fetuși cu sindrom Potter .
Caracteristicile neonatale ale sindromului Potter includ modificări faciale, malformații ale membrelor, limitare a creșterii fetale și hipoplazie pulmonară, cunoscute sub numele de tetradă oligohidramnios. Aceste trăsături apar ca urmare a compresiei fetale datorate oligohidramniosului prelungit. Faciesul Potter se caracterizează prin hipertelorism, pleoapă mongolă, pliu epicanthic, punte nazală și lobul urechii aplatizate, nas în cioc de papagal, urechi joase, bărbie retrasă, un mic pliu sub buză, gât scurt și lanțuri suplimentare în jurul gâtului. Prezentarea clinică a bebelușului din acest studiu a fost fața retrasă, punte nazală și lobul urechii aplatizate, nas în cioc de papagal, urechi joase, bărbie mică, scurtă și retrasă, un pliu mic sub buză gât scurt și pliuri suplimentare în jurul gâtului, mâini largi, deviere a mâinii la încheietura mâinii, valgus bilateral și limitare severă a creșterii fetale, ceea ce a dus la o greutate la naștere de 800 g. Aceste manifestări clinice au fost în concordanță cu Shastry și Al-Haggar .
Presiunea constantă a pereților uterini asupra peretelui toracic al fătului și presiunea organelor intraabdominale asupra diafragmului constituie una dintre consecințele oligohidramniosului în sindromul Potter, care sunt, de asemenea, principalele cauze ale hipoplaziei și insuficienței pulmonare în acest sindrom. Gravitatea hipoplaziei pulmonare depinde de faza de dezvoltare a plămânului în care apare oligohidramniosul, precum și de intensitatea și durata oligohidramniosului. Din cauza detresei respiratorii severe și a hipoplaziei pulmonare, fetușii se nasc cu rinichi displazici sau mor la scurt timp după naștere . În acest studiu, indicele de lichid amniotic a fost zero conform ultrasonografiei, iar nou-născutul a avut o detresă respiratorie severă la naștere și a murit la câteva momente după naștere.
Sindromul Potter poate fi asociat cu anomalii cardiace congenitale, anomalii gastrointestinale (cum ar fi atrezia esofagului, agenezia colonului și malformațiile anale și duodenale, diverticulul lui Meckel și chisturile pancreatice și ale splinei), tulburări scheletice, anomalii cerebrale și diverse asociații, cum ar fi Vakarl et al. În studiul de față, ecografia a raportat o cardiomegalie severă (mai mult de 80%) și vezica urinară invizibilă; iar examenul fizic al nou-născutului a evidențiat anomalii ale membrelor și un anus imperforat.
Cauza principală a sindromului Potter rămâne neclară în majoritatea cazurilor, dar are un motiv genetic în unele cazuri, iar modelul de moștenire depinde de o anumită cauză genetică. Anomaliile genetice, cum ar fi moștenirea autosomal dominantă sau recesivă a bolii polichistice renale, displazia renală ereditară, cauzată de mutații ale genelor RET și UPK3A și anomalii cromozomiale, pot duce la anomalii de dezvoltare și pot duce la sindromul Potter. Acest sindrom apare sporadic, dar poate fi moștenit atunci când provine din triada autosomal dominantă. Sindromul Potter este mai frecvent la copiii care au un istoric familial de anomalii renale . În studiul de față nu a fost găsită nicio cauză specifică pentru această boală și nu au existat antecedente familiale de anomalii renale. San și Samal au subliniat, de asemenea, etiologia necunoscută a sindromului Potter și motivele genetice ale acestuia în studiile lor .
Chiar dacă principala cauză a sindromului Potter este anomalia în dezvoltarea rinichilor, diagnosticul inițial se realizează prin ecografie care poate arăta pierderea sau absența lichidului amniotic și absența vezicii urinare, și este continuat prin investigații privind prezența sau absența rinichilor. Consilierea genetică este importantă pentru a confirma diagnosticul. Identificarea acestui sindrom, care se caracterizează prin agenezie renală bilaterală, hipoplazie pulmonară, facies Potter și malformații ale membrelor, poate fi problematică prin ecografie din cauza oligohidramniosului . În acest studiu, ecografia a evidențiat absența indicelui de lichid amniotic, invizibilitatea vezicii urinare și agenezia renală bilaterală.
Sindromul Potter este incompatibil cu viața și are un prognostic mortal. Sarcina poate fi întreruptă înainte ca fătul să atingă stadiul de viață. Îngrijirea standard a sarcinii nu se schimbă atunci când se decide continuarea sarcinii . Una dintre limitările studiului de față a fost că nu s-au efectuat teste genetice, deoarece părinții nu au acceptat autopsia.
În studiul de față, o mamă aflată în a 25-a săptămână de sarcină cu un făt cu sindrom Potter, diagnosticat prin ecografie, a fost internată în secția de nașteri și a născut normal un copil care a murit la câteva momente după naștere.
Concluzie
Sindromul Potter este o boală foarte gravă și o afecțiune mortală. Ecografia prenatală ajută la depistarea acestui sindrom prin examinarea oligohidramniosului și a afecțiunilor renale.
Conflict de interese
Autorii declară că nu au niciun conflict de interese în ceea ce privește publicarea acestui raport de caz.
Finanțare
Nici o finanțare nu a fost solicitată sau obținută în legătură cu acest raport de caz.
Consimțământul pacientului
Obținut
Proveniența și evaluarea colegială
Acest raport de caz a fost evaluat de colegi.
- Potter EL. Caracteristicile faciale ale sugarilor cu agenezie renală bilaterală. Am J Obstet Gynecol 1946; 51: 885-888.
- Srihari Babu M, Asha Latha D, Radhika P. Sindromul Potters: Un raport de caz. JDMS 2015; 14: 14: 14-16.
- Sudhanshu Kumar D, Sidharth Sankar M. Potter facies cu boală polichistică de rinichi în asociere cu alte anomalii congenitale: două rapoarte de caz. J Med Dent Sci 2013; 2: 2665-2668.
- Khatami F. Sindromul Potters: Un studiu de 15 pacienți. Arch Iranian Med 2004; 7: 186- 189.
- Sarkar S, Das Gupta S, Barua M, Ghosh R, Mondal K, Chatterjee U, Datta C. Secvența Potters: O poveste a celor mai rare, mai rare și mai rare. Indian J Pathol Microbiol 2015; 58: 102-104.
- Manoj MG, Kakkar S. Sindromul Potters – O constelație fatală de anomalii. Indian J Med Res 2014; 139: 648-649.
- Himabindu A, Narasinga B. Un caz fatal de sindrom Potters-un raport de caz. JCDR 2011; 5: 1264-1266.
- Moreira BL, Monarim MA, Romano RF, Mattos LA, DIppolito G. Sindromul Doege-Potter. Radiol Bras 2015; 48: 195-196.
- Reuvers JR, van Dorp M, Van Schil PE. Tumoare fibroasă solitară a pleurei cu sindrom Doege-Potter asociat. Acta Chir Belg 2016; 116: 386-387.
- Shastry SM, Kolte SS, Sanagapati PR. Secvența Potters. J Clin Neonatol 2012; 1: 157-159.
- Al-Haggar M, Yahia S, Abdel-Hadi D, Grill F, Al-Kaissi A. Sirenomelia (symelia apus) cu sindromul Potters în legătură cu diabetul zaharat gestațional: Un raport de caz și o revizuire a literaturii. Afr Health Sci 2010; 10: 395-399.
- Dhundiraj KM, Madhukar DN, Ambadasrao PG, Wamanrao KS, Prem ZM. Sindromul Potters: Un raport de 5 cazuri. Indian J Pathol Microbiol 2006; 49: 254-257.
- Dayal J, Maheshwari E, Ghai PS, Bhatotia S. Diagnosticul antenatal cu ultrasunete al sindromului Potters. Indian J Radiol Imaging 2003; 13: 81-83.
- San Agustin JT, Klena N, Granath K, Panigrahy A, Stewart E, Devine W, Strittmatter L, Jonassen JA, Liu X. Legătura genetică între defectele renale din naștere și boala cardiacă congenitală. Nat Commun 2016; 22: 111-118.
- Samal SK, Rathod S. Sirenomelia: Sindromul sirenei: Raportul a două cazuri. J Nat Sci Biol Med 2015; 6: 264-266.
- Gerards FA, Twisk JW, Fetter WP. Predicția hipoplaziei pulmonare cu ultrasonografie 2 sau 3-dimensională în sarcini complicate. Am J Obstet Gynecol 2008; 198: 140-146.
- Kemper MJ, Mueller-Wiefel DE. Prognosis of antenatally diagnosed oligohydramnios of renal origin. Eur J Pediatr 2007; 166: 393-398.