Case Report – Biomedical Research (2019) Volume 30, Issue 2
Report of a deadly case of potter syndrome: Un rapporto di caso.
Mehrbano Amirshahi, Mahin Badakhsh* e Zohreh Sadat Hashemi
Dipartimento di ostetricia, School of Nursing and Midwifery, Zabol University of Medical sciences, Zabol, Iran
*Corresponding Author: Mahin Badakhsh
Dipartimento di Ostetricia
Scuola di Infermieristica e Ostetricia
Zabol University of Medical sciences
Iran
Data accettata: February 21, 2019
DOI: 10.35841/biomedicalresearch.30-19-084
Visita per altri articoli correlati a Biomedical Research
Abstract
Background: La sindrome di Potter è un raro disordine congenito che si riferisce ad un insieme di manifestazioni cliniche che sono associate con oligoidramnios causato da insufficienza renale fetale. Il segno distintivo di questa sindrome è il particolare quadro clinico che oltre all’oligoidramnios caratterizza con ipoplasia polmonare, agenesia renale bilaterale; deformità degli arti e faccia specifica. L’embrione muore prima o subito dopo la nascita per insufficienza respiratoria. Lo scopo di questo studio è quello di segnalare un feto con la sindrome di Potter che è nato da un parto vaginale. Rapporto del caso: L’esame ecografico della donna incinta ha rivelato che il suo feto maschio con età gestazionale di 25 settimane con la sindrome di Potter e l’indice di liquido amniotico è zero. La madre è stata ricoverata in travaglio unitario e ha partorito con un decorso naturale. Il bambino aveva un quadro clinico della sindrome di Potter e grave distress respiratorio ed è morto poco dopo la nascita. Conclusione: La sindrome di Potter è una condizione molto grave e il più delle volte è mortale. L’ecografia prenatale esaminando l’oligoidramnios e i reni aiuta a fare la diagnosi.
Parole chiave
Sindrome di Potter, Oligoidramnios, Ipoplasia polmonare, Feto
Introduzione
La sindrome di Potter è una rara malformazione congenita che colpisce principalmente i feti maschi ed è caratterizzata da ipoplasia polmonare causata da insufficienza renale. È stata segnalata per la prima volta da Edith Potter nel 1946. Dopo la 16a settimana di gravidanza, la quantità di liquido amniotico dipende principalmente dalla produzione di urina da parte del feto. Durante la vita intrauterina, il feto inghiotte continuamente il liquido amniotico che ritorna nel sacco amniotico tramite i reni. L’oligoidramnios si verifica quando il volume del liquido amniotico è inferiore al normale per quel periodo della gravidanza. Questa diminuzione di volume può essere dovuta a una riduzione della produzione di urina, secondaria a cause come l’agenesia renale bilaterale, l’ostruzione del tratto urinario e la rottura prolungata dei sacchi amniotici. L’urina del feto, essenziale per lo sviluppo dei polmoni, svolge il suo ruolo contribuendo allo sviluppo delle vie aeree e degli alveoli, creando pressione idraulica e fornendo prolina, un aminoacido essenziale per lo sviluppo dei polmoni. Se gli alveoli e di conseguenza i polmoni non sono adeguatamente sviluppati alla nascita, il neonato non sarà in grado di respirare bene e soffrirà di distress respiratorio a causa dell’ipoplasia polmonare. Pertanto, secondaria all’insufficienza renale, l’ipoplasia polmonare è la principale causa di morte nei neonati con la sindrome di Potter.
La produzione di urina da parte dell’embrione non solo influenza il volume del liquido amniotico, ma preserva anche l’embrione contro la pressione della parete dell’utero della madre come un cuscino. L’oligoidramnios provoca una forma speciale di feto chiamata “Potter facies”, che è caratterizzata da caratteristiche come il ponte nasale appiattito, il mento ipoplasico, le pieghe epicantali, la cataratta e le orecchie basse.
La causa principale della sindrome di Potter è sconosciuta; questa sindrome ha un background genetico in alcuni casi, ed è più comune nei neonati con una storia familiare di anomalie renali. La sindrome, con un’incidenza di 1 ogni 2.000-5.000 feti, è associata ad un rischio di ricorrenza del 3-6%, e si trova nello 0,2-0,4% delle autopsie in neonati morti o che muoiono subito dopo la nascita. Tuttavia, si ritiene che la malattia possa avere una prevalenza maggiore perché i feti colpiti nascono morti o muoiono poco dopo la nascita. Non esiste un metodo conosciuto per prevenire questa malattia mortale. Pertanto, l’ecografia di screening è raccomandata a 16-18 settimane di gestazione per le madri con una storia positiva di gravidanza per questa sindrome, al fine di valutare l’oligoidramnios e i reni fetali. Anche se questa sindrome ha conseguenze mortali e non è compatibile con la vita e termina con la morte del bambino, i feti affetti richiedono la rianimazione e il trattamento dell’ostruzione dell’uscita urinaria al momento del parto. Il presente studio ha voluto segnalare un feto con la sindrome di Potter che è nato attraverso un parto normale.
Rapporto del caso
Il caso era un feto maschio di 25 settimane con la sindrome di Potter, nato attraverso un parto normale. La madre è stata ammessa al reparto maternità dell’ospedale Amir al-Momenin, Zabol, Iran, il 10 maggio 2017 con un’ecografia, che ha mostrato l’assenza di liquido amniotico e la diagnosi di sindrome di Potter per il feto. La madre aveva 24 anni, iraniana, casalinga, gravida 3, e para 1, e aveva una storia di aborto al mese 1. Non ricordava il primo giorno della sua ultima mestruazione, e la sua età gestazionale era di 25 settimane secondo l’unica ecografia eseguita lo stesso giorno. Aveva una storia di parto normale (il suo figlio precedente era sano e aveva avuto un parto normale), e aveva ricevuto cure prenatali durante la gravidanza. I suoi test di gravidanza erano normali. Non c’erano precedenti di fumo, droghe o alcol, malattie di base come il diabete, l’ipertensione, la tiroide, cardiopolmonare e renale, infezioni, e uso di farmaci diversi da quelli di routine della gravidanza (integratori di ferro e vitamine). La madre e suo marito erano cugini e lei non ha menzionato una storia familiare di una nascita neonatale con un disturbo simile. I test sierologici prenatali erano negativi per sifilide, AIDS, epatite B e rosolia. Aveva un gruppo sanguigno B positivo.
Secondo l’ecografia menzionata, l’età gestazionale era di 25 settimane, l’indice del liquido amniotico era zero, i reni del feto erano invisibili nella normale posizione anatomica, la vescica era invisibile, e c’era una grave restrizione della crescita fetale e una grave cardiomegalia (più di 80%). The diagnosis was Potter syndrome based on ultrasound, after which the mother was taken to the maternity ward.
The mother was hospitalized and, after the normal course of labor, gave birth to a male new-born through vaginal method. The boy had growth limitation (800 g birth weight), the clinical presentation of Potter syndrome, and severe respiratory distress and died immediately after delivery (Figures 1 and 2).
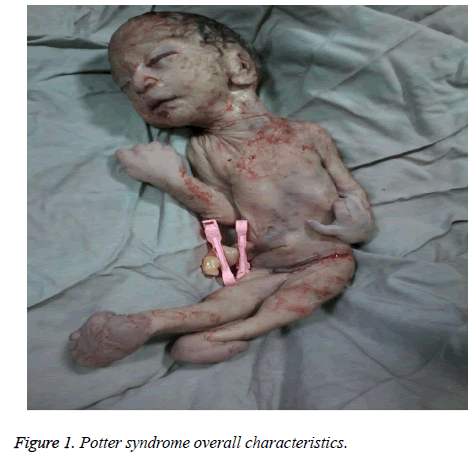
Figure 1: Potter syndrome overall characteristics.
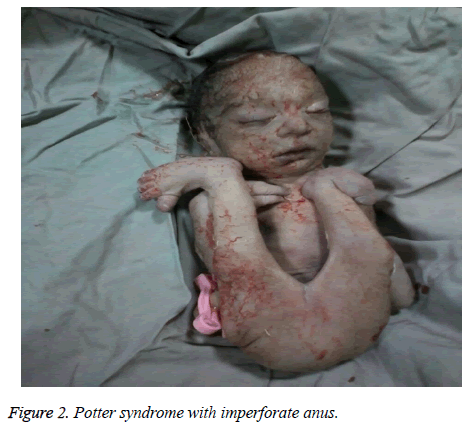
Figure 2: Potter syndrome with imperforate anus.
Discussion
The present study introduced a rare case that has not been reported in the region so far. It was a fatal case of Potter syndrome that was born through normal delivery. L’ecografia della donna ha rivelato un feto a 25 settimane di gravidanza con sindrome di Potter e un indice di liquido amniotico pari a zero. La madre è stata ricoverata in sala parto e il bambino è nato per via vaginale. Il neonato con la presentazione clinica della sindrome di Potter aveva una grave sofferenza respiratoria ed è morto poco dopo la nascita.
La sindrome di Potter, descritta per la prima volta da Edith Potter nei neonati, è caratterizzata da agenesia renale bilaterale o altre anomalie renali come aplasia, displasia, ipoplasia o malattia renale policistica. È riconosciuto più nei ragazzi con un rapporto maschio-femmina di 2 a 1, suggerendo la presenza di alcuni geni sul cromosoma Y . La sindrome di Potter può essere vista in neonati con reni normali, ma la madre aveva una perdita cronica e prolungata di liquido amniotico durante le settimane centrali della gravidanza.
In questo studio, l’ecografia delle donne incinte ha rivelato un caso di sindrome di Potter in un feto maschio alla 25° settimana di gravidanza, un indice di liquido amniotico nullo, e la mancanza di entrambi i reni e della vescica. I risultati ecografici dei feti con gravi malattie renali in 23 famiglie hanno indicato un oligoidramnios prolungato, una grave disfunzione renale e la facies di Potter, e i bambini sono morti entro ore o giorni dopo la nascita. Moreira e Reuvers hanno anche sottolineato la nascita di feti con la sindrome di Potter.
Le caratteristiche neonatali della sindrome di Potter includono alterazioni facciali, malformazioni degli arti, limitazione della crescita fetale e ipoplasia polmonare, nota come oligoidramnios tetrad. Queste caratteristiche derivano dalla compressione fetale dovuta all’oligoidramnios prolungato. La facies di Potter è caratterizzata da ipertelorismo, palpebra mongola, piega epicantica, ponte nasale e lobo dell’orecchio appiattiti, naso a becco di pappagallo, orecchie basse, mento incassato, una piccola piega sotto il labbro, collo corto e catene extra intorno al collo. La presentazione clinica del neonato in questo studio era faccia incassata, ponte nasale e lobo dell’orecchio appiattiti, naso a becco di pappagallo, orecchie basse, mento piccolo, corto e incassato, una piccola piega sotto il labbro, collo corto e pieghe extra intorno al collo, mani larghe, deviazione della mano al polso, valgismo bilaterale e grave limitazione della crescita fetale, che ha portato ad un peso alla nascita di 800 g. Queste manifestazioni cliniche erano coerenti con Shastry e Al-Haggar.
La pressione costante delle pareti uterine sulla parete toracica del feto e la pressione degli organi intra-addominali sul diaframma costituiscono una delle conseguenze dell’oligoidramnios nella sindrome di Potter, che sono anche le cause principali dell’ipoplasia e del fallimento polmonare in questa sindrome. La gravità dell’ipoplasia polmonare dipende dalla fase di sviluppo del polmone in cui si verifica l’oligoidramnios, così come l’intensità e la durata dell’oligoidramnios. A causa del grave distress respiratorio e dell’ipoplasia polmonare, i feti nascono con reni displastici o muoiono poco dopo la nascita. In questo studio, l’indice del liquido amniotico era zero secondo l’ecografia e il neonato aveva una grave sofferenza respiratoria alla nascita ed è morto pochi istanti dopo la nascita.
La sindrome di Potter può essere associata ad anomalie cardiache congenite, anomalie gastrointestinali (come atresia dell’esofago, agenesia del colon e malformazioni anali e duodenali, diverticolo di Meckel e cisti del pancreas e della milza), disturbi scheletrici, anomalie del cervello e varie associazioni, come Vakarl et al. Nel presente studio, l’ecografia ha riportato una grave cardiomegalia (più dell’80%) e una vescica invisibile; e l’esame fisico del neonato ha rivelato anomalie degli arti e un ano imperforato.
La causa principale della sindrome di Potter rimane poco chiara nella maggior parte dei casi, ma ha una ragione genetica in alcuni casi, e il modello di eredità dipende da una particolare causa genetica. Anomalie genetiche come l’ereditarietà autosomica dominante o recessiva della malattia policistica del rene, la displasia renale ereditaria, causata da mutazioni del gene RET e UPK3A e anomalie cromosomiche, possono provocare anomalie di sviluppo e portare alla sindrome di Potter. Questa sindrome si verifica sporadicamente ma può essere ereditata quando nasce dalla triade autosomica dominante. La sindrome di Potter è più comune nei bambini che hanno una storia familiare di anomalie renali. Nessuna causa specifica è stata trovata nel presente studio per questa malattia, e non c’era nessuna storia familiare di anomalie renali. San e Samal hanno anche sottolineato l’eziologia sconosciuta della sindrome di Potter e le sue ragioni genetiche nei loro studi.
Anche se la causa principale della sindrome di Potter è l’anomalia nello sviluppo dei reni, la diagnosi iniziale viene effettuata tramite ecografia che può mostrare la perdita o l’assenza di liquido amniotico e l’assenza di vescica, e viene continuata tramite indagini sulla presenza o assenza di reni. La consulenza genetica è importante per confermare la diagnosi. L’identificazione di questa sindrome, che è caratterizzata da agenesia renale bilaterale, ipoplasia polmonare, facies Potter, e malformazione degli arti, può essere problematico da ecografia a causa di oligoidramnios . In questo studio, l’ecografia ha mostrato l’assenza di indice di liquido amniotico, l’invisibilità della vescica e l’agenesia renale bilaterale.
La sindrome di Potter è incompatibile con la vita e ha una prognosi mortale. La gravidanza può essere interrotta prima che il feto raggiunga lo stadio vitale. La cura standard della gravidanza non cambia quando si decide di continuare la gravidanza. Una delle limitazioni del presente studio è che non sono stati eseguiti test genetici, poiché i genitori non hanno accettato l’autopsia.
Nel presente studio, una madre alla 25° settimana di gravidanza con un feto con sindrome di Potter, diagnosticata tramite ecografia, è stata ricoverata in unità di parto e ha dato un parto normale a un bambino che è morto pochi istanti dopo la nascita.
Conclusione
La sindrome di Potter è una malattia molto grave e una condizione mortale. L’ecografia prenatale aiuta a rilevare questa sindrome esaminando l’oligoidramnios e le condizioni dei reni.
Conflitto di interesse
Gli autori dichiarano di non avere alcun conflitto di interesse riguardo alla pubblicazione di questo case report.
Finanziamento
Nessun finanziamento è stato richiesto o assicurato in relazione a questo rapporto di caso.
Consenso del paziente
Ricevuto
Provenienza e Peer Review
Questo rapporto di caso è stato sottoposto a peer review.
- Potter EL. Caratteristiche facciali dei neonati con agenesia renale bilaterale. Am J Obstet Gynecol 1946; 51: 885-888.
- Srihari Babu M, Asha Latha D, Radhika P. Sindrome di Potters: A case report. JDMS 2015; 14: 14-16.
- Sudhanshu Kumar D, Sidharth Sankar M. Potter facies con malattia renale policistica in associazione con altre anomalie congenite: due case report. J Med Dent Sci 2013; 2: 2665-2668.
- Khatami F. Potters syndrome: Uno studio di 15 pazienti. Arch Iranian Med 2004; 7: 186- 189.
- Sarkar S, Das Gupta S, Barua M, Ghosh R, Mondal K, Chatterjee U, Datta C. Potters sequenza: Una storia del raro, più raro e più raro. Indian J Pathol Microbiol 2015; 58: 102-104.
- Manoj MG, Kakkar S. Potters syndrome-A fatal constellation of anomalies. Indian J Med Res 2014; 139: 648-649.
- Himabindu A, Narasinga B. Un caso fatale di sindrome di Potters-un rapporto di caso. JCDR 2011; 5: 1264-1266.
- Moreira BL, Monarim MA, Romano RF, Mattos LA, DIppolito G. Doege-Potter syndrome. Radiol Bras 2015; 48: 195-196.
- Reuvers JR, van Dorp M, Van Schil PE. Tumore fibroso solitario della pleura con associata la sindrome di Doege-Potter. Acta Chir Belg 2016; 116: 386-387.
- Shastry SM, Kolte SS, Sanagapati PR. Sequenza di Potters. J Clin Neonatol 2012; 1: 157-159.
- Al-Haggar M, Yahia S, Abdel-Hadi D, Grill F, Al-Kaissi A. Sirenomelia (symelia apus) con sindrome di Potters in connessione con il diabete mellito gestazionale: Un rapporto di caso e revisione della letteratura. Afr Health Sci 2010; 10: 395-399.
- Dhundiraj KM, Madhukar DN, Ambadasrao PG, Wamanrao KS, Prem ZM. Sindrome di Potters: Un rapporto di 5 casi. Indian J Pathol Microbiol 2006; 49: 254-257.
- Dayal J, Maheshwari E, Ghai PS, Bhatotia S. diagnosi ecografica prenatale della sindrome di Potters. Indian J Radiol Imaging 2003; 13: 81-83.
- San Agustin JT, Klena N, Granath K, Panigrahy A, Stewart E, Devine W, Strittmatter L, Jonassen JA, Liu X. Collegamento genetico tra difetti di nascita renali e malattie cardiache congenite. Nat Commun 2016; 22: 111-118.
- Samal SK, Rathod S. Sirenomelia: The mermaid syndrome: report of two cases. J Nat Sci Biol Med 2015; 6: 264-266.
- Gerards FA, Twisk JW, Fetter WP. Predire l’ipoplasia polmonare con ecografia 2 o 3-dimensionale in gravidanze complicate. Am J Obstet Gynecol 2008; 198: 140-146.
- Kemper MJ, Mueller-Wiefel DE. Prognosis of antenatally diagnosed oligohydramnios of renal origin. Eur J Pediatr 2007; 166: 393-398.