Relato de caso – Pesquisa Biomédica (2019) Volume 30, Edição 2
Relatório de um caso mortal de síndrome de Oleiro: Relato de um caso.
Mehrbano Amirshahi, Mahin Badakhsh* e Zohreh Sadat Hashemi
Departamento de Obstetrícia, Escola de Enfermagem e Obstetrícia, Universidade Zabol de Ciências Médicas, Zabol, Irão
Autor correspondente: Mahin Badakhsh
Departamento de Obstetrícia
Escola de Enfermagem e Obstetrícia
Universidade de Ciências Médicas de Zabol
Irão
Data aceite: 21 de fevereiro de 2019
DOI: 10.35841/biomedicalresearch.30-19-084
Visita para mais artigos relacionados na Biomedical Research
Abstract
Background: A síndrome de Potter é uma doença congênita rara que se refere a um conjunto de manifestações clínicas que estão associadas a oligoidrâmnios causados por insuficiência renal fetal. A marca desta síndrome é o quadro clínico especial que além de oligoidrâmnios característico com hipoplasia pulmonar, agenesia renal bilateral; deformidades de membros e face específica. O embrião morre antes ou imediatamente após o nascimento devido a insuficiência respiratória. O objetivo deste estudo é relatar um feto com síndrome de Potter que nasceu por parto vaginal. Relato de caso: O exame ultra-sonográfico da mulher grávida revelou que o feto masculino com idade gestacional de 25 semanas com síndrome de Potter e o índice de líquido amniótico é zero. A mãe foi hospitalizada em trabalho de parto unitário e deu à luz em um curso natural. O bebê tinha um quadro clínico de síndrome de Potter e grave angústia respiratória e morreu logo após o nascimento. Conclusão: A síndrome de Potter é uma condição muito grave e, na maioria das vezes, é mortal. A ultra-sonografia pré-natal através do exame de oligoidrâmnios e rins ajuda a diagnosticar.
Síndrome de Potter, Oligoidrâmnios, Hipoplasia pulmonar, Feto
Introdução
Síndrome de Potter é uma malformação congênita rara que afeta principalmente os fetos masculinos e é caracterizada por hipoplasia pulmonar causada por insuficiência renal. Foi relatada pela primeira vez por Edith Potter em 1946. Após a 16ª semana de gravidez, a quantidade de líquido amniótico depende principalmente da produção de urina pelo feto. Durante a vida intra-uterina, o feto engole continuamente o líquido amniótico que retorna ao saco amniótico pelos rins. Os oligoidrâmnios ocorrem quando o volume de líquido amniótico é inferior ao normal durante esse período de gravidez. Esta diminuição no volume pode ser devida a uma redução no débito urinário, secundária a causas como agenesia renal bilateral, obstrução do trato urinário e ruptura prolongada dos sacos amnióticos. A urina fetal, essencial para o desenvolvimento dos pulmões, desempenha seu papel de contribuir para o desenvolvimento das vias aéreas e alvéolos, criando pressão hidráulica e fornecendo prolina, um aminoácido essencial para o desenvolvimento dos pulmões. Se os alvéolos e como resultado os pulmões não estiverem adequadamente desenvolvidos ao nascer, o neonato não será capaz de respirar bem e sofrerá de angústia respiratória devido à hipoplasia pulmonar. Portanto, secundária à insuficiência renal, a hipoplasia pulmonar é a principal causa de morte em neonatos com síndrome de Potter .
Produção de urina pelo embrião não só afeta o volume de líquido amniótico, mas também preserva o embrião contra a pressão da parede uterina da mãe como uma almofada. Oligohydramnios resulta em uma forma especial de feto chamada “Potter facies”, que é caracterizada por características como ponte nasal achatada, queixo hipoplásico, dobras epicantais, catarata e orelhas baixas .
A principal causa da síndrome de Potter é desconhecida; esta síndrome tem um fundo genético em alguns casos, e é mais comum em neonatos com história familiar de anormalidades renais . A síndrome, com uma incidência de 1 em cada 2.000 a 5.000 fetos, está associada a um risco de recorrência de 3-6%, e é encontrada em 0,2-0,4% das autópsias em recém-nascidos mortos ou naqueles que morrem imediatamente após o nascimento . No entanto, acredita-se que a doença possa ter uma prevalência mais elevada porque os fetos afectados nascem mortos ou morrem pouco tempo após o nascimento. Não existe um método conhecido para prevenir esta doença mortal. Portanto, recomenda-se a ultra-sonografia de rastreio às 16-18 semanas de gestação para mães com uma história positiva de gravidez para esta síndrome, com o objectivo de avaliar oligoidrâmnios e rins fetais. Embora esta síndrome tenha consequências mortais e não seja compatível com a vida e termine com a morte do bebé, os fetos afectados requerem ressuscitação e tratamento da obstrução do débito urinário no momento do parto. O presente estudo teve como objetivo relatar um feto com síndrome de Potter que nasceu através do parto normal.
Caso
O caso foi um feto masculino de 25 semanas com síndrome de Potter, nascido através do parto normal. A mãe foi internada na maternidade do Hospital Amir al-Momenin, Zabol, Irã, em 10 de maio de 2017, com ultra-som, que mostrou a ausência de líquido amniótico e o diagnóstico de síndrome de Potter para o feto. A mãe tinha 24 anos, Iraniana, dona de casa, grávida 3, e para 1, e tinha um histórico de aborto no Mês 1. Ela não se lembrava do primeiro dia de sua última menstruação, e sua idade gestacional era de 25 semanas de acordo com a única ultrassonografia realizada no mesmo dia. Ela tinha uma história de parto normal (seu filho anterior era saudável e tinha tido parto normal), e tinha recebido atendimento pré-natal durante a gravidez. Os testes de gravidez dela eram normais. Não havia histórico de tabagismo, drogas ou álcool, doenças subjacentes como diabetes, hipertensão, tiróide, cardiopulmonar e renal, infecções e uso de outras drogas além dos medicamentos de gravidez de rotina (suplementos de ferro e vitaminas). A mãe e o marido eram primos e ela não mencionou um histórico familiar de um parto neonatal com um distúrbio semelhante. Os testes serológicos pré-natais foram negativos para sífilis, AIDS, hepatite B e rubéola. Ela tinha um tipo sanguíneo B positivo.
De acordo com o ultra-som mencionado, a idade gestacional foi de 25 semanas, o índice de líquido amniótico foi zero, os rins do feto eram invisíveis na posição anatômica normal, a bexiga era invisível, e havia restrição de crescimento fetal grave e cardiomegalia grave (mais de 80%). The diagnosis was Potter syndrome based on ultrasound, after which the mother was taken to the maternity ward.
The mother was hospitalized and, after the normal course of labor, gave birth to a male new-born through vaginal method. The boy had growth limitation (800 g birth weight), the clinical presentation of Potter syndrome, and severe respiratory distress and died immediately after delivery (Figures 1 and 2).
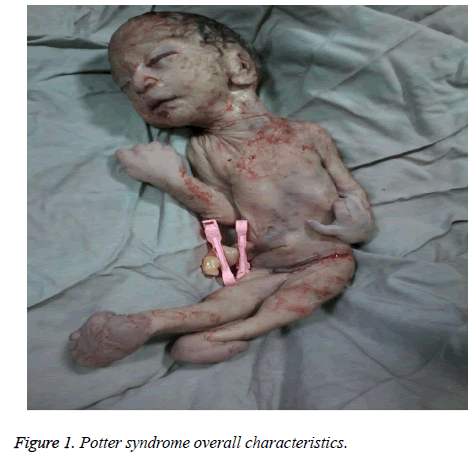
Figure 1: Potter syndrome overall characteristics.
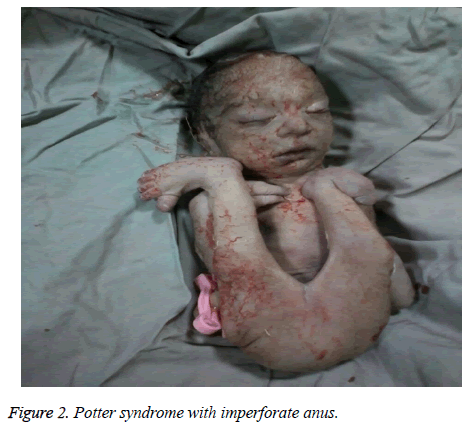
Figure 2: Potter syndrome with imperforate anus.
Discussion
The present study introduced a rare case that has not been reported in the region so far. It was a fatal case of Potter syndrome that was born through normal delivery. A ultrassonografia da mulher revelou um feto às 25 semanas de gestação com síndrome de Potter e um índice de líquido amniótico de zero. A mãe foi admitida na unidade de parto e o bebé nasceu vaginalmente. O recém-nascido com a apresentação clínica da síndrome de Potter teve grave angústia respiratória e morreu logo após o nascimento.
Síndrome de Potter, primeiramente descrita por Edith Potter em recém-nascidos, é caracterizada por agenesia renal bilateral ou outras anormalidades renais como aplasia, displasia, hipoplasia ou doença renal policística. É mais reconhecida em meninos com proporção de 2 para 1 entre homens e mulheres, sugerindo a presença de certos genes no cromossomo Y . A síndrome de Potter pode ser vista em neonatos com rins normais, mas a mãe teve um vazamento crônico e prolongado de líquido amniótico durante as semanas médias de gravidez .
Neste estudo, a ultrassonografia das gestantes revelou um caso de síndrome de Potter em um feto masculino na 25ª semana de gravidez, um índice de líquido amniótico zero, e a falta tanto de rins quanto de bexiga. Os achados ultrassonográficos de fetos com doença renal grave em 23 famílias indicaram oligoidrâmnio prolongado, disfunção renal grave e fácies de Potter, e os bebês morreram de horas a dias após o nascimento. Moreira e Reuvers também apontaram o nascimento de fetos com síndrome de Potter .
As características neonatais da síndrome de Potter incluem alterações faciais, malformações de membros, limitação do crescimento fetal e hipoplasia pulmonar, conhecida como oligoidrâmnio tetrad. Estas características surgem da compressão fetal devido a oligoidramnetos prolongados. Potter facies é caracterizado por hipertelorismo, pálpebra mongol, prega epicanética, ponte nasal achatada e lóbulo da orelha, nariz de bico de papagaio, orelhas baixas, queixo recuado, uma pequena prega abaixo do lábio, pescoço curto, e correntes extras ao redor do pescoço. A apresentação clínica da criança neste estudo foi: face recuada, ponte nasal achatada e lobo das orelhas, nariz com bico de papagaio, orelhas baixas, queixo pequeno, curto e recuado, uma pequena prega abaixo do lábio pescoço curto, e pregas extras ao redor do pescoço, mãos largas, desvio da mão no pulso, valgo bilateral e limitação severa do crescimento fetal, o que resultou em um peso ao nascer de 800 g. Estas manifestações clínicas foram consistentes com Shastry e Al-Haggar .
A pressão constante das paredes uterinas na parede torácica do feto e a pressão dos órgãos intra-abdominais no diafragma constituem uma das consequências dos oligoidrâmnios na síndrome de Potter, que são também as principais causas de hipoplasia e falência pulmonar nesta síndrome. A gravidade da hipoplasia pulmonar depende da fase de desenvolvimento do pulmão em que ocorre o oligoidrâmnio, bem como da intensidade e duração do oligoidrâmnio. Devido à grave angústia respiratória e hipoplasia pulmonar, os fetos nascem com rins displásicos ou morrem logo após o nascimento. Neste estudo, o índice de líquido amniótico foi zero de acordo com a ultra-sonografia e o recém-nascido teve grave desconforto respiratório ao nascer e morreu alguns momentos após o nascimento.
Síndrome de Potter pode estar associada a anomalias cardíacas congênitas, anomalias gastrointestinais (como atresia do esôfago, agenesia colônica, malformações anais e duodenais, divertículo de Meckel e cistos pancreáticos e do baço), distúrbios esqueléticos, anormalidades cerebrais e várias associações, como Vakarl et al. No presente estudo, a ultrassonografia relatou uma cardiomegalia grave (mais de 80%) e bexiga invisível; e o exame físico do neonato revelou anomalias de membros e um ânus imperfurado.
A principal causa da síndrome de Potter permanece pouco clara na maioria dos casos, mas tem uma razão genética em alguns casos, e o padrão de herança depende de uma causa genética particular. Anormalidades genéticas, como herança autossômica dominante ou recessiva de doença renal policística, displasia renal hereditária, causada por mutações dos genes RET e UPK3A e anormalidades cromossômicas, podem resultar em anormalidades de desenvolvimento e levar à síndrome de Potter. Esta síndrome ocorre esporadicamente, mas pode ser herdada quando originada da tríade autossômica dominante. A síndrome de Potter é mais comum em bebês que têm uma história familiar de anormalidades renais. Nenhuma causa específica foi encontrada no presente estudo para esta doença, e não houve história familiar de anomalias renais. San e Samal também apontaram a etiologia desconhecida da síndrome de Potter e suas razões genéticas em seus estudos .
Embora a principal causa da síndrome de Potter seja a anormalidade no desenvolvimento renal, o diagnóstico inicial é feito por ultra-som que pode mostrar a perda ou ausência de líquido amniótico e a ausência de bexiga, e é continuado através de investigação sobre a presença ou ausência de rins. O aconselhamento genético é importante para confirmar o diagnóstico. A identificação desta síndrome, que é caracterizada por agenesia renal bilateral, hipoplasia pulmonar, fácies de Potter e malformação de membros, pode ser problemática pela ultra-sonografia devido a oligoidrâmnio. Neste estudo, a ultrassonografia mostrou a ausência de índice de líquido amniótico, invisibilidade da bexiga e agenesia renal bilateral.
Síndrome de Potter é incompatível com a vida e tem um prognóstico mortal. A gravidez pode ser interrompida antes do feto atingir a fase de vida. Os cuidados padrão de gravidez não mudam quando se decide continuar a gravidez. Uma das limitações do presente estudo foi que não foram realizados testes genéticos, pois os pais não aceitaram a autópsia.
No presente estudo, uma mãe na sua 25ª semana de gravidez com um feto com síndrome de Potter, diagnosticada através de ultra-som, foi admitida na unidade de parto e deu um parto normal a um bebê que morreu alguns momentos após o nascimento.
Conclusão
Síndrome de Potter é uma doença muito grave e uma condição mortal. A ultrassonografia pré-natal ajuda a detectar essa síndrome, examinando oligoidrâmnios e condições renais.
Conflito de interesse
Os autores declaram não ter conflito de interesse em relação à publicação deste relato de caso.
Funding
Nenhum financiamento foi solicitado ou assegurado em relação a este relato de caso.
Consentimento Paciente
Bemtido
Proveniência e Revisão pelos Pares
Este relato de caso foi revisto por pares.
- Potter EL. Características faciais de bebês com agenesia renal bilateral. Am J Obstet Gynecol 1946; 51: 885-888.
- Srihari Babu M, Asha Latha D, Síndrome de Radhika P. Potters: Um relato de caso. JDMS 2015; 14: 14-16.
- Sudhanshu Kumar D, Sidharth Sankar M. Potter facies com doença renal policística em associação com outras anomalias congênitas: dois relatos de caso. J Med Dent Sci 2013; 2: 2665-2668.
- Sarkar S, Das Gupta S, Barua M, Ghosh R, Mondal K, Chatterjee U, Datta C. Sequência de Potters: Uma história do mais raro, mais raro e mais raro. Indian J Pathol Microbiol 2015; 58: 102-104.
- Himabindu A, Narasinga B. Um caso fatal de síndrome de Potters – um relato de caso. JCDR 2011; 5: 1264-1266.
- Moreira BL, Monarim MA, Romano RF, Mattos LA, DIppolito G. Doege-Potter syndrome. Radiol Bras 2015; 48: 195-196.
- Reuvers JR, van Dorp M, Van Schil PE. Tumor fibroso solitário da pleura com síndrome de Doege-Potter associada. Acta Chir Belg 2016; 116: 386-387.
- Shastry SM, Kolte SS, Sanagapati PR. Sequência de oleiros. J Clin Neonatol 2012; 1: 157-159.
- Al-Haggar M, Yahia S, Abdel-Hadi D, Grill F, Al-Kaissi A. Sirenomelia (symelia apus) com síndrome de Potters em conexão com a diabetes mellitus gestacional: Um relato de caso e revisão de literatura. Afr Health Sci 2010; 10: 395-399.
- Dhundiraj KM, Madhukar DN, Ambadasrao PG, Wamanrao KS, Prem ZM. Síndrome de Potters: Um relatório de 5 casos. Indian J Pathol Microbiol 2006; 49: 254-257.
- Dayal J, Maheshwari E, Ghai PS, Bhatotia S. Diagnóstico ultra-sonográfico pré-natal da síndrome de Potters. Indian J Radiol Imaging 2003; 13: 81-83.
- San Agustin JT, Klena N, Granath K, Panigrahy A, Stewart E, Devine W, Strittmatter L, Jonassen JA, Liu X. Genetic link between renal birth defects and congenital heart disease. Nat Commun 2016; 22: 111-118.
- Gerards FA, Twisk JW, Fetter WP. Previsão de hipoplasia pulmonar com ultra-sonografia bidimensional ou tridimensional em gestações complicadas. Am J Obstet Gynecol 2008; 198: 140-146.
- Kemper MJ, Mueller-Wiefel DE. Prognosis of antenatally diagnosed oligohydramnios of renal origin. Eur J Pediatr 2007; 166: 393-398.
li> Khatami F. Síndrome de Potters: Um estudo de 15 pacientes. Arch Iranian Med 2004; 7: 186- 189.
li> Manoj MG, Kakkar S. Potters syndrome-A fatal constellation of anomalies. Indian J Med Res 2014; 139: 648-649.
li> Samal SK, Rathod S. Sirenomelia: A síndrome da sereia: relato de dois casos. J Nat Sci Biol Med 2015; 6: 264-266.