Wprowadzenie
Zespół regresji ogonowej (CRS) jest nieczęstym zaburzeniem opisanym po raz pierwszy przez Geoffroya Saint-Hilaire’a i Hohla w 1852 r., a w 1964 r. Duhmel ukuł termin „zespół regresji ogonowej”.1-3 CRS jest zaburzeniem spowodowanym anomalią dystalnych segmentów kręgosłupa i obejmuje szeroki zakres anomalii, takich jak częściowa agenezja rdzenia kręgowego, związane z nią wady rozwojowe miednicy, niedomykalność odbytu, wady rozwojowe narządów płciowych, anomalie serca, obustronna dysplazja lub aplazja nerek, hipoplazja płuc oraz skrajna rotacja zewnętrzna z fuzją stawów dolnych, której najcięższą postacią jest sirenomelia (zespół syreny). CRS wiąże się również z hipoplazją kości udowej, deformacją stóp i przykurczem zgięciowym kończyn dolnych. Inteligencja jest ogólnie zachowana.1,2,4 dotyczy od 0,1 do 0,25 na każde 10 000 ciąż, a stosunek mężczyzn do kobiet wynosi 2,7:1.3-5 Na poziomie embrionalnym uważa się, że CRS jest wynikiem defektów w indukcji elementów ogonowych w zarodku przed 28 dniem ciąży. Uszkodzenie jest wytwarzane w tylnej przyśrodkowej osi mezodermy, powodując brak rozwoju mezoblastycznego żółtka ogoniastego.4,6 Dokładna etiologia nie jest znana; jednakże cukrzyca matki, predyspozycje genetyczne i hipoperfuzja naczyniowa zostały zasugerowane jako możliwe czynniki.4-7
Cukrzyca przedciążowa jest bez wątpienia teratogenem i istnieją dowody, że cukrzyca ciążowa może mieć wpływ na rozwój najcięższej postaci CRS.5
Pinter i Reece udowodnili, że zmiany wywołane hiperglikemią w zamykaniu cewy nerwowej obejmują nieuporządkowane komórki, zmniejszenie mitozy i zmiany wskazujące na przedwczesne dojrzewanie. Metabolizm oksydacyjny zmieniony przez cukrzycę matki może powodować wzrost produkcji wolnych rodników tlenowych w rozwijającym się zarodku, co może być teratogenne.8 Od 16% do 22% przypadków CRS jest związanych z cukrzycą matki, co zwiększa ryzyko urodzenia dziecka z CRS nawet o 400.3,4,7 Opisano kilka przypadków rodzin z CRS, co sugeruje możliwość transmisji genetycznej z różnymi możliwymi sposobami transmisji: X-linked dominant, multi-factorial polygenic, and autosomal dominant patterns with reduced penetrance and variable expressivity.7 The „vascular theft” theory was initially proposed by Kampmeier in 1927 and re-introduced in 1986 by Stevenson. Adra i wsp. uznali teorię „kradzieży naczyniowej” Stevensona za możliwą etiologię patologii CRS.7 Na etapie rozwoju embrionalnego bardziej ogoniaste struktury są oddzielone od elementów głowowych, takich jak mózg, kręgosłup i rdzeń kręgowy, stąd brak zmian poznawczych w tym zespole.4
Wyróżnia się 2 grupy CRS: pierwsza grupa jest najbardziej dotknięta chorobą, z zakończeniem rdzenia kręgowego powyżej L1. Kość krzyżowa kończy się na S1, a w niektórych przypadkach jest nieobecna. Pacjenci z drugiej grupy wykazują mniej nasiloną dysgenezję z niską implantacją rdzenia kręgowego, skrępowaną pogrubiałym filum terminale lub tłuszczakiem wewnątrzrdzeniowym.4
Prezentacja przypadku
Przedstawiamy przypadek noworodka z 40-letnią matką z historią medyczną nieleczonych włókniaków macicy, nieregularnymi cyklami ginekologicznymi, 3 ciążami z 2 porodami i cholecystektomią w 2008 roku. Kobieta wspomina o ciąży nieplanowanej, ale chcianej, z opieką prenatalną w ciąży wysokiego ryzyka. Uczestniczyła w ponad 15 konsultacjach prenatalnych. W 18. tygodniu ciąży stwierdzono zgodne dane o wadach rozwojowych cewy nerwowej z agenezją kości krzyżowej, co skutkowało rozpoznaniem CRS. W drugim trymestrze ciąży stwierdzono hiperglikemię, w związku z czym pacjentka została objęta leczeniem insuliną. W 40 tygodniu ciąży wykonano cięcie cesarskie uzyskując produkt zdrowy, z punktacją w skali Apgar 8/9 przy wieku ciążowym 39,6 tygodnia. Podczas badania fizykalnego zaobserwowano wagę 3,44 kg (7,58 lbs), długość 49 cm (19,2 in), obwód głowy 35 cm (13,77 in) i obwód brzucha 32 cm (12,59 in). Podczas oględzin zaobserwowano ewidentną fetopatię cukrzycową, z obfitym owłosieniem i niską implantacją (ryc. 1). Normalne ciemiączko przednie (2 x 2 cm), twarz okrągła, wydatne kości policzkowe, poziome bruzdy podniebienne, krótki mostek nosowy, okrągły czubek, cienkie wargi, pełne podniebienie, dysplazja kosteczek słuchowych z hipertrichozą małżowin usznych, krótka szyja z grzbietowym wybrzuszeniem, klatka piersiowa prawidłowa, bez szmerów przy osłuchiwaniu, brzuch miękki, bez wyczuwalnych mas i trzewioczaszki, kręgosłup linijny z obecnością wgłębienia na skórze w okolicy lędźwiowo-krzyżowej (ryc. 2), brak kości krzyżowej. 2), nie wyczuwano kości krzyżowej, odbyt był prawidłowo położony, drożny, z brakiem tonusu. Stwierdzono wyraźne skrócenie kończyn dolnych oraz obustronną stopę koślawą (ryc. 3). Wykonano zdjęcia rentgenowskie w celu uwidocznienia wad kostnych, stwierdzając całkowitą agenezję kości krzyżowej z zespoleniem lędźwiowo-biodrowym. Badanie rezonansu magnetycznego mózgu i kręgosłupa (MRI) (ryc. 4) wykazało przerwanie cięciwy i zakończenie odcinka lędźwiowego na poziomie L3. Za tym poziomem możemy zidentyfikować jedynie bezpostaciowe masy o intensywności sygnału tłuszczowego, związane z tkanką podskórną. Krótki rdzeń kręgowy, płaski conus medullaris na poziomie T10, obie kości biodrowe hipoplastyczne, stopione na poziomie środkowym. Nie jesteśmy w stanie zobaczyć kości krzyżowej. Widoczne były obie nerki malrotacyjne i hipoplastyczne, pęcherz moczowy wydatny, zgodny z pęcherzem neurogennym. Obraz mózgu był prawidłowy. W celu wykluczenia innych anomalii trzewnych zlecono wykonanie echografii górnego piętra jamy brzusznej, stwierdzając dyskretną rozbieżność w wielkości lewej nerki, dodatkowo nie stwierdzając jej prawidłowej konfiguracji, co sugerowało dyskretną hipoplazję, a także możliwą malrotację. W trakcie pobytu przeprowadzono konsultację wielospecjalistyczną, założono szyny gipsowe na obie kończyny dolne. Pacjent pozostaje w obserwacji urologicznej, traumatologicznej, neurologicznej i ogólnopediatrycznej, co ma na celu zwrócenie uwagi na wszystkie czynniki ryzyka i ewentualne powikłania.
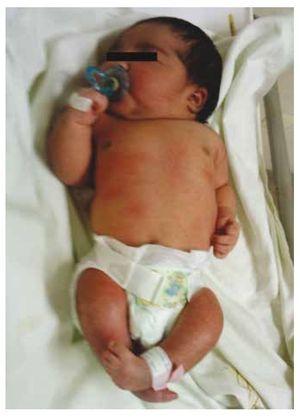
Rycina 1 Noworodek z fetopatią cukrzycową.
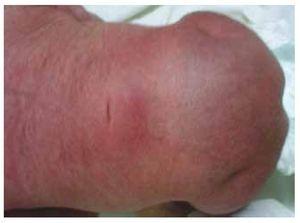
Rysunek 2 Wgłębienie na skórze w okolicy lędźwiowo-krzyżowej.
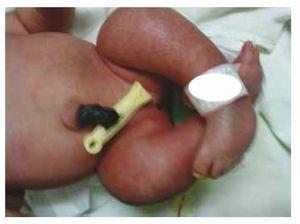
Figure 3 Equinovarus feet.
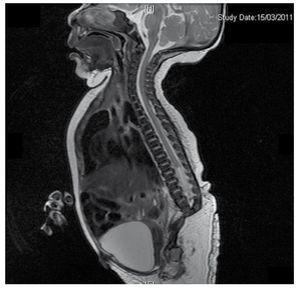
Figure 4 The lumbar spine magnetic resonance imaging (MRI) shows an abrupt chord and lumbar termination at the L3 level. Short spinal cord, flat conus medullaris at T10 level, hypoplastic iliac bones, fused at mid-level. No sacrum is seen.
Discussion
The CRS is a rare congenital malformation. Even though the specific etiologic factor is unknown, it is related to maternal diabetes, genetic predisposition and vascular hypoperfusion.3 Just as in the case presented, this alteration is characterized by agenesis of the sacrum involving iliac and lumbar vertebrae with their corresponding spinal segments and variable abnormalities in the lower limbs as well as in other organs. Fetal diagnosis tools allow early syndrome detection. Najczęściej stosowanym narzędziem paraklinicznym jest prenatalne badanie ultrasonograficzne, którego kluczowym elementem jest szczegółowa ocena kręgosłupa i kończyn dolnych; pozwala ono również na rozpoznanie CRS poprzez wykazanie nagłego zakończenia odcinka lędźwiowego kręgosłupa i hipoplastycznych kończyn dolnych. Podczas stawiania diagnozy prenatalnej należy skupić się na określeniu stopnia digenezji oraz towarzyszących jej wad wrodzonych w celu ustalenia rokowania i zaplanowania w odpowiednim czasie postnatalnych interwencji terapeutycznych.3,5 Klasyfikacja Renshava, która powstała w 1978 roku, dzieli zespół na 4 stopnie w zależności od nasilenia agenezji kości krzyżowej oraz zajęcia kręgów biodrowych i lędźwiowych.9 Według tej klasyfikacji nasz pacjent należy do stopnia iV (całkowita agenezja kości krzyżowej z zespoleniem kości biodrowych). Ta grupa wiąże się z jeszcze gorszym rokowaniem, większym obciążeniem neurologicznym i następstwami wielonarządowymi, głównie na poziomie nerek. Opisany przypadek jest dobitnym przykładem szerokiego spektrum zmian, jakie mogą wystąpić u rozwijającego się płodu w wyniku niekontrolowanej cukrzycy matki w czasie ciąży. Ze względu na wysoką korelację między tą wadą a matką chorą na cukrzycę i jej rozwojem we wczesnym okresie ciąży, konieczne jest zastosowanie strategii zapobiegawczej, obejmującej ścisłą kontrolę glikemii przed okresem organogenezy zarodka, a nawet wcześniej u pacjentek z grupy wysokiego ryzyka. Ważne jest również odpowiednie poradnictwo i przedkoncepcyjne badania genetyczne. Leczenie jest wyzwaniem zarówno dla lekarza, jak i dla rodziców i wymaga wielodyscyplinarnego podejścia z udziałem pediatry, chirurga dziecięcego, ortopedy, fizykoterapeuty i urologa, w zależności od stopnia zaawansowania choroby. Given the fact that the primary pathology is irreversible, treatment is just supportive, with the sole purpose of accomplishing a life as normal as possible.
Conflicts of interest
The authors have no conflicts of interest to declare.
Funding
No financial support was provided.
Received: September 2013;
Accepted: January 2014