Introdução
Síndrome de regressão caudal (SRC) é um distúrbio pouco frequente descrito pela primeira vez por Geoffroy Saint-Hilaire e Hohl em 1852, e em 1964 Duhmel cunhou o termo “síndrome de regressão caudal”.1-3 O SRC é uma desordem causada por uma anomalia dos segmentos distais da coluna vertebral, e se estende a uma ampla gama de anomalias como agenesia parcial da medula espinhal, malformações pélvicas associadas, ânus imperfurado, malformações genitais, anomalias cardíacas, displasia ou aplasia renal bilateral, hipoplasia pulmonar e rotação externa extrema com fusão das articulações inferiores resultando na forma mais grave em sirenomelia (síndrome de sereia). A SRC também está associada à hipoplasia femoral, pés deformados e contração da flexão das extremidades inferiores. A inteligência é preservada, em geral.1,2,4 afeta entre 0,1 e 0,25 a cada 10.000 gestações, com uma relação macho-fêmea de 2,7:1,3-5 A nível embrionário, acredita-se que o SRC é o resultado de defeitos na indução de elementos caudais no embrião antes do 28º dia de gestação. A lesão é produzida no eixo mesodérmico medial posterior causando a ausência de desenvolvimento da gema mesoblástica caudal.4,6 A etiologia exata é desconhecida; entretanto, diabetes materno, predisposição genética e hipoperfusão vascular têm sido sugeridos como possíveis fatores.4-7
A diabetes pré-gestacional é sem dúvida um teratógeno, e há boas evidências de que a diabetes gestacional pode estar implicada no desenvolvimento da forma mais grave de CRS.5
Pinter e reece provaram que alterações induzidas pela hiperglicemia no fechamento do tubo neural incluem células desordenadas, diminuição da mitose e alterações que indicam uma maturação prematura. O metabolismo oxidativo alterado pela diabetes materna pode causar um aumento na produção de radicais livres de oxigênio no embrião em desenvolvimento, que pode ser teratogênico.8 Entre 16% e 22% dos casos de SRC estão associados à diabetes mellitus materna, o que aumenta o risco de ter um filho com SRC em até 400,3,4,7 Vários casos de famílias com SRC foram relatados, o que sugere uma possível transmissão genética com diferentes modos de transmissão possíveis: Padrões poligênicos multifatoriais dominantes, multifatoriais e autossômicos com penetração reduzida e expressividade variável.7 A teoria do “roubo vascular” foi inicialmente proposta por Kampmeier em 1927 e re-introduzida em 1986 por Stevenson. Adra et al. consideraram a teoria do “roubo vascular” de Stevenson como uma possível etiologia da patologia do SRC.7 Durante o estágio de desenvolvimento embrionário, as estruturas mais caudais são separadas dos elementos cefálicos como o cérebro, a coluna vertebral e a medula espinhal, daí a ausência de alterações cognitivas nesta síndrome.4
Existem 2 grupos de SRC: o primeiro grupo é o mais afetado com a terminação da medula espinhal acima de L1. O sacro termina em S1, e em alguns casos, está ausente. Os pacientes do segundo grupo apresentam uma disgênese menos grave com baixa implantação da medula espinhal e amarrados por um filamento terminal espessado ou lipoma intraespinhal.4
Apresentação do caso
Apresentamos o caso de uma recém-nascida de uma mãe de 40 anos com história médica de fibróides uterinos não tratados, histórico de ciclos ginecológicos irregulares, 3 gestações com 2 partos e coleistectomia em 2008. Ela menciona uma gravidez não planejada mas desejada, com uma gravidez de alto risco pré-natal. Ela assistiu a mais de 15 consultas pré-natais. Na 18ª semana de gestação, foram encontrados dados compatíveis com malformações do tubo neural com agenesia sacral, resultando em um diagnóstico de SRC. Durante o segundo trimestre de gestação foi detectada hiperglicemia, portanto ela foi colocada em tratamento com insulina. Com 40 semanas de gestação, foi realizada a cesariana obtendo-se um produto saudável, com pontuação de Apgar de 8/9, com idade gestacional de 39,6 semanas. No exame físico observou-se um peso de 3,44 kg (7,58 lbs), um comprimento de 49 cm (19,2 in), uma circunferência da cabeça de 35 cm (13,77 in) e uma circunferência abdominal de 32 cm (12,59 in). Durante a inspeção pudemos observar uma evidente fetopatia diabética, com abundância de pêlos e baixa implantação (Fig. 1). Fontanela anterior normal (2 x 2 cm), face redonda, maçãs do rosto proeminentes, fissuras palpebrais horizontais, ponte nasal curta, ponta arredondada, lábios finos, palato cheio, displasia dos pavilhões auriculares com hipertricose da hélice, pescoço curto com abaulamento dorsal, tórax normal, sem sopros durante a auscultação, abdômen mole sem massas palpáveis ou visceromegalia, coluna vertebral linear com presença de covinha na pele na região lombossacral (Fig. 2), nenhum osso sacro era palpável, posição normal do ânus, permeável, com tônus ausente. Houve encurtamento evidente das extremidades inferiores e do pé torto bilateral varo (Fig. 3). Realizaram-se radiografias para ver as malformações ósseas e pudemos observar a agenesia completa do sacro com uma fusão lombo-ilíaca. Uma ressonância magnética do cérebro e coluna vertebral (RM) (Fig. 4), relatou um acorde abrupto e terminação lombar ao nível L3. Após este nível conseguimos identificar apenas massas amorfas com intensidade de sinal de gordura, relacionadas ao tecido subcutâneo. Medula espinhal curta, cono medular plana no nível T10, ambos os ossos ilíacos estavam hipoplásicos, e fundidos no nível médio. Não conseguimos ver o sacro. Foi possível ver ambos os rins mal-rotados e hipoplásicos, uma bexiga urinária proeminente compatível com uma bexiga neurogénica. A imagem do cérebro foi relatada como normal. Foi solicitada uma ecografia do abdómen superior para excluir outras anomalias viscerais, encontrando uma discrepância no tamanho do rim esquerdo, para além de não encontrar a sua configuração normal, o que sugere uma hipoplasia discreta, bem como uma possível mal-rotação. Durante a sua estadia realizámos uma consulta multidisciplinar, tendo sido colocadas talas de gesso em ambos os membros inferiores. O paciente permanece em acompanhamento com Urologia, Traumatologia, Neurologia e Pediatria Geral, isto com o objetivo de manter um acompanhamento atento a todos os fatores de risco e possíveis complicações.
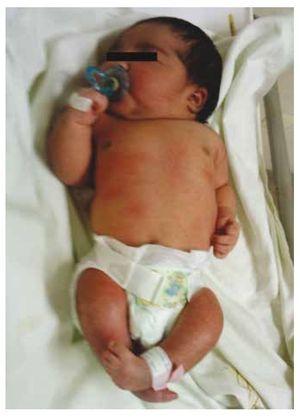
Figure 1 Newborn with diabetic fetopathy.
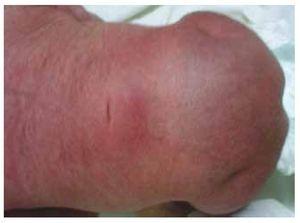
Figure 2 A dimple on the skin at the lumbosacral region.
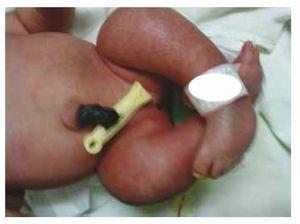
Figure 3 Equinovarus feet.
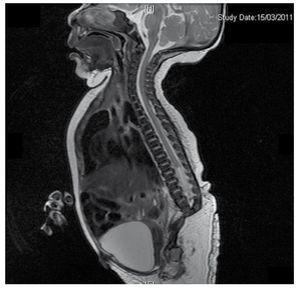
Figure 4 The lumbar spine magnetic resonance imaging (MRI) shows an abrupt chord and lumbar termination at the L3 level. Short spinal cord, flat conus medullaris at T10 level, hypoplastic iliac bones, fused at mid-level. No sacrum is seen.
Discussion
The CRS is a rare congenital malformation. Even though the specific etiologic factor is unknown, it is related to maternal diabetes, genetic predisposition and vascular hypoperfusion.3 Just as in the case presented, this alteration is characterized by agenesis of the sacrum involving iliac and lumbar vertebrae with their corresponding spinal segments and variable abnormalities in the lower limbs as well as in other organs. Fetal diagnosis tools allow early syndrome detection. A ultra-sonografia pré-natal é o instrumento paraclínico mais utilizado; um elemento chave da ultra-sonografia pré-natal é a avaliação detalhada da coluna vertebral e dos membros inferiores; também permite um diagnóstico CRS através da demonstração de uma terminação abrupta da coluna lombar e dos membros inferiores hipoplásicos. Ao fazer um diagnóstico pré-natal, devemos nos concentrar em discernir o grau de digenesia, bem como as anomalias congênitas associadas, com o objetivo de estabelecer um prognóstico e um plano oportuno de intervenções terapêuticas pós-natais.3,5 A classificação de renshav, criada em 1978, categoriza a síndrome em 4 graus com base na gravidade da agenesia do sacro e envolvimento das vértebras ilíacas e lombares.9 Segundo esta classificação, nosso paciente pertence ao grau iV (agenesia completa do sacro com fusão ilíaca dos ossos). Este grupo está associado a um prognóstico ainda pior, com maior impacto neurológico e seqüelas multissistêmicas, principalmente a nível renal. Este caso é um exemplo claro da ampla gama de alterações que podem afetar o feto em crescimento como resultado de diabetes materno descontrolado durante a gravidez. Devido à alta correlação entre este defeito e a mãe diabética, e seu desenvolvimento durante os estágios iniciais da gravidez, é imperativo ter uma estratégia preventiva que inclua um rigoroso controle glicêmico antes do período de organogênese embrionária, ou mesmo antes, em pacientes de alto risco. Uma orientação adequada e exames genéticos pré-gestacionais também são importantes. O tratamento é um desafio tanto para o médico quanto para os pais, e exige uma abordagem multidisciplinar envolvendo um pediatra, um cirurgião pediátrico, um cirurgião ortopédico, um fisioterapeuta e um urologista, dependendo da gravidade. Given the fact that the primary pathology is irreversible, treatment is just supportive, with the sole purpose of accomplishing a life as normal as possible.
Conflicts of interest
The authors have no conflicts of interest to declare.
Funding
No financial support was provided.
Received: September 2013;
Accepted: January 2014