Takeaways
-
En mjukvara för ”djupinlärning” från det Google-ägda labbet DeepMind har gjort stora framsteg när det gäller att lösa en av biologins största utmaningar – att förstå hur proteiner viks.
-
Proteinveckning är den process genom vilken ett protein tar sin form från en sträng av byggstenar till sin slutliga tredimensionella struktur, som bestämmer dess funktion.
-
Om forskare bättre kan förutsäga hur proteiner tar sin struktur, eller ”veckar sig”, kan de snabbare utveckla läkemedel som till exempel blockerar verkan av viktiga virusproteiner.
Att lösa det som biologer kallar ”problemet med proteinets veckning” är en stor sak. Proteiner är cellernas arbetshästar och finns i alla levande organismer. De består av långa kedjor av aminosyror och är viktiga för cellernas struktur och kommunikationen mellan dem samt för att reglera all kemi i kroppen.
Denna vecka demonstrerade det Google-ägda företaget för artificiell intelligens DeepMind ett djupinlärningsprogram kallat AlphaFold2, som experter kallar ett genombrott för att lösa den stora utmaningen med proteinveckning.
Proteiner är långa kedjor av aminosyror som länkas samman som pärlor på ett snöre. Men för att ett protein ska kunna göra sitt jobb i cellen måste det ”veckas” – en process av vridning och böjning som förvandlar molekylen till en komplex tredimensionell struktur som kan interagera med sitt mål i cellen. Om veckningen störs kommer proteinet inte att få rätt form – och det kommer inte att kunna utföra sitt arbete i kroppen. Detta kan leda till sjukdom – vilket är fallet med en vanlig sjukdom som Alzheimers och sällsynta sjukdomar som cystisk fibros.
Djupinlärning är en beräkningsteknik som använder den ofta dolda information som finns i stora datamängder för att lösa frågor av intresse. Den har använts flitigt inom områden som spel, tal- och röstigenkänning, autonoma bilar, vetenskap och medicin.
Jag tror att verktyg som AlphaFold2 kommer att hjälpa forskare att utforma nya typer av proteiner, sådana som till exempel kan hjälpa till att bryta ner plast och bekämpa framtida viruspandemier och sjukdomar.
Jag är en beräkningskemist och författare till boken The State of Science. Mina studenter och jag studerar strukturen och egenskaperna hos fluorescerande proteiner med hjälp av datorprogram för proteinveckning som bygger på klassisk fysik.
Efter årtionden av studier av tusentals forskargrupper är dessa program för prediktion av proteinveckning mycket bra på att beräkna strukturella förändringar som sker när vi gör små ändringar i kända molekyler.
Men de har inte lyckats tillräckligt bra med att förutsäga hur proteiner viks från grunden. Innan djupinlärning kom fram verkade proteinveckningsproblemet omöjligt svårt, och det verkade vara redo att frustrera beräkningskemister i många decennier framöver.
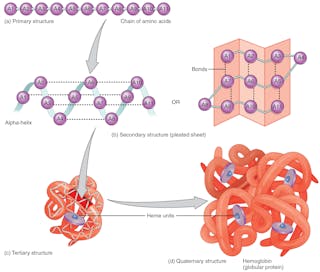
Proteinveckning
Aminosyrornas sekvens – som är kodad i DNA – definierar proteinets 3D-form. Formen bestämmer dess funktion. Om proteinets struktur förändras kan det inte utföra sin funktion. Att korrekt förutsäga proteinveckningar baserat på aminosyrasekvensen skulle kunna revolutionera utformningen av läkemedel och förklara orsakerna till nya och gamla sjukdomar.
Alla proteiner med samma sekvens av aminosyrabyggstenar veckar sig till samma tredimensionella form, vilket optimerar interaktionerna mellan aminosyrorna. De gör detta inom millisekunder, trots att de har ett astronomiskt antal möjliga konfigurationer tillgängliga för dem – ungefär 10 gånger 300. Det är detta enorma antal som gör det svårt att förutsäga hur ett protein veckas även när forskarna känner till hela sekvensen av aminosyror som ingår i dess tillverkning. Tidigare var det omöjligt att förutsäga proteinets struktur utifrån aminosyrasekvensen. Proteinstrukturer måste bestämmas experimentellt, ett tidskrävande och dyrt arbete.
När forskarna bättre kan förutsäga hur proteiner veckas kan de bättre förstå hur cellerna fungerar och hur felveckade proteiner orsakar sjukdomar. Bättre verktyg för att förutsäga proteiner kommer också att hjälpa oss att utforma läkemedel som kan rikta sig mot en viss topologisk region i ett protein där kemiska reaktioner äger rum.

AlphaFold är sprungen ur djuplärande schack-, Go- och pokerspel
Framgången för DeepMinds program för att förutsäga proteinveckning, kallat AlphaFold, är inte oväntad. Andra djupinlärningsprogram skrivna av DeepMind har demolerat världens bästa schack-, Go- och pokerspelare.
2016 blev Stockfish-8, en schackmotor med öppen källkod, världsmästare i datorschack. Den utvärderade 70 miljoner schackställningar per sekund och hade århundraden av ackumulerade mänskliga schackstrategier och årtionden av datorvana att ta tillvara på. Den spelade effektivt och brutalt och slog skoningslöst alla sina mänskliga utmanare utan ett uns av finess. Inför djupinlärning.
Den 7 december 2017 besegrade Googles djupinlärande schackprogram AlphaZero Stockfish-8. Schackmaskinerna spelade 100 partier, där AlphaZero vann 28 och spelade 72 oavgjorda partier. Det förlorade inte ett enda parti. AlphaZero gjorde bara 80 000 beräkningar per sekund, jämfört med Stockfish-8:s 70 miljoner beräkningar, och det tog bara fyra timmar att lära sig schack från grunden genom att spela mot sig själv några miljoner gånger och optimera sina neurala nätverk allteftersom den lärde sig av sina erfarenheter.
AlphaZero lärde sig ingenting av människor eller schackspel som spelades av människor. Den lärde sig själv och i processen kom den fram till strategier som aldrig tidigare setts. I en kommentar i tidskriften Science skrev den tidigare schackvärldsmästaren Garry Kasparov att genom att lära sig av att spela själv utvecklade AlphaZero strategier som ”återspeglar sanningen” om schack snarare än att återspegla ”programmerarnas prioriteringar och fördomar”. ”Det är förkroppsligandet av klyschan ’arbeta smartare, inte hårdare’.”
CASP – OS för molekylmodellerare
Vartannat år testar världens främsta beräkningskemister förmågan hos sina program att förutsäga proteinernas veckning och tävlar i tävlingen Critical Assessment of Structure Prediction (CASP).
I tävlingen får lagen den linjära sekvensen av aminosyror för cirka 100 proteiner för vilka 3D-formen är känd men ännu inte publicerad; de måste sedan beräkna hur dessa sekvenser skulle veckas. År 2018 slog AlphaFold, den djupinlärande nykomlingen i tävlingen, alla traditionella program – men knappt.
Två år senare, i måndags, tillkännagavs det att AlphaFold2 hade vunnit 2020 års tävling med god marginal. Det piskade sina konkurrenter, och dess förutsägelser var jämförbara med befintliga experimentella resultat som fastställts med hjälp av guldstandardtekniker som röntgendiffraktionskristallografi och kryoelektronmikroskopi. Snart förväntar jag mig att AlphaFold2 och dess avkomma kommer att vara de metoder som väljs för att bestämma proteinstrukturer innan man tillgriper experimentella tekniker som kräver mödosamt och arbetsamt arbete med dyra instrument.
En av anledningarna till AlphaFold2:s framgång är att den kunde använda sig av proteindatabasen, som har över 170 000 experimentellt bestämda 3D-strukturer, för att träna sig själv på att beräkna korrekt veckade strukturer av proteiner.
Den potentiella effekten av AlphaFold kan uppskattas om man jämför antalet av alla publicerade proteinstrukturer – cirka 170 000 – med de 180 miljoner DNA- och proteinsekvenser som deponerats i Universal Protein Database. AlphaFold kommer att hjälpa oss att sortera bland skattkistor av DNA-sekvenser på jakt efter nya proteiner med unika strukturer och funktioner.
Har AlphaFold gjort mig, en molekylärmodellerare, överflödig?
Som med schack- och Go-programmen – AlphaZero och AlphaGo – vet vi inte exakt vad AlphaFold2-algoritmen gör och varför den använder sig av vissa korrelationer, men vi vet att den fungerar.
Förutom att hjälpa oss att förutsäga strukturer för viktiga proteiner kommer förståelsen av AlphaFolds ”tänkande” också att hjälpa oss att få nya insikter i mekanismen för proteinveckning.
En av de vanligaste farhågorna som uttrycks om artificiell intelligens är att den kommer att leda till storskalig arbetslöshet. AlphaFold har fortfarande en betydande väg att gå innan den konsekvent och framgångsrikt kan förutsäga proteinveckning.
När det väl har mognat och programmet kan simulera proteinveckning kommer dock beräkningskemister att vara integrerat involverade i att förbättra programmen, försöka förstå de underliggande korrelationer som används och tillämpa programmet för att lösa viktiga problem, t.ex. den felveckning av proteiner som är förknippad med många sjukdomar, t.ex. Alzheimers sjukdom, Parkinsons sjukdom, cystisk fibros och Huntingtons sjukdom.
AlphaFold och dess avkomma kommer säkerligen att förändra arbetssättet för beräkningskemister, men det kommer inte att göra dem överflödiga. Andra områden kommer inte att vara lika lyckligt lottade. Tidigare kunde robotar ersätta människor som utförde manuellt arbete; med artificiell intelligens utmanas även våra kognitiva färdigheter.