Fallbericht – Biomedizinische Forschung (2019) Band 30, Ausgabe 2
Bericht über einen tödlichen Fall des Potter-Syndroms: Ein Fallbericht.
Mehrbano Amirshahi, Mahin Badakhsh* und Zohreh Sadat Hashemi
Department of Midwifery, School of Nursing and Midwifery, Zabol University of Medical Sciences, Zabol, Iran
*Korrespondierender Autor: Mahin Badakhsh
Department of Midwifery
School of Nursing and Midwifery
Zabol University of Medical sciences
Iran
Accepted date: February 21, 2019
DOI: 10.35841/biomedicalresearch.30-19-084
Besuchen Sie für weitere verwandte Artikel bei Biomedical Research
Abstract
Hintergrund: Das Potter-Syndrom ist eine seltene angeborene Störung, die sich auf eine Reihe von klinischen Manifestationen bezieht, die mit einer durch fetales Nierenversagen verursachten Oligohydramnion verbunden sind. Kennzeichnend für dieses Syndrom ist das besondere klinische Bild, das zusätzlich zum Oligohydramnion charakteristisch ist für pulmonale Hypoplasie, bilaterale Nierenagenesie, Gliedmaßendeformitäten und ein spezifisches Gesicht. Der Embryo stirbt vor oder unmittelbar nach der Geburt aufgrund von Atemversagen. Ziel dieser Studie ist es, über einen Fötus mit Potter-Syndrom zu berichten, der vaginal geboren wurde. Fallbericht: Bei der Ultraschalluntersuchung der schwangeren Frau wurde festgestellt, dass ihr männlicher Fötus mit einem Gestationsalter von 25 Wochen das Potter-Syndrom aufwies und der Fruchtwasserindex null betrug. Die Mutter wurde mit Wehen ins Krankenhaus eingeliefert und entband auf natürlichem Wege. Das Kind wies das klinische Bild eines Potter-Syndroms und schwere Atemnot auf und starb kurz nach der Geburt. Schlussfolgerung: Das Potter-Syndrom ist eine sehr ernste Erkrankung, die in den meisten Fällen tödlich verläuft. Ein pränataler Ultraschall mit Untersuchung von Oligohydramnion und Nieren hilft bei der Diagnose.
Schlüsselwörter
Potter-Syndrom, Oligohydramnion, Lungenhypoplasie, Fötus
Einführung
Das Potter-Syndrom ist eine seltene angeborene Fehlbildung, die vor allem männliche Föten betrifft und durch eine Lungenhypoplasie mit Nierenversagen gekennzeichnet ist. Das Syndrom wurde erstmals 1946 von Edith Potter beschrieben. Nach der 16. Schwangerschaftswoche hängt die Menge des Fruchtwassers hauptsächlich von der Urinproduktion des Fötus ab. Während des intrauterinen Lebens schluckt der Fötus ständig Fruchtwasser, das von den Nieren in die Fruchtblase zurückgeführt wird. Oligohydramnion tritt auf, wenn das Fruchtwasservolumen geringer ist als normal für diesen Zeitraum der Schwangerschaft. Diese Abnahme des Volumens kann auf eine verringerte Urinausscheidung zurückzuführen sein, die durch Ursachen wie beidseitige Nierenagenesie, Harnwegsobstruktion und anhaltende Ruptur der Fruchtblase bedingt ist. Der Urin des Fötus, der für die Entwicklung der Lungen unerlässlich ist, trägt zur Entwicklung der Atemwege und der Lungenbläschen bei, erzeugt hydraulischen Druck und liefert Prolin, eine für die Entwicklung der Lungen unerlässliche Aminosäure. Wenn die Alveolen und damit die Lungen bei der Geburt nicht ausreichend entwickelt sind, kann das Neugeborene nicht gut atmen und leidet aufgrund der Lungenhypoplasie unter Atemnot. Daher ist die Lungenhypoplasie neben dem Nierenversagen die Haupttodesursache bei Neugeborenen mit Potter-Syndrom.
Die Urinproduktion des Embryos wirkt sich nicht nur auf das Fruchtwasservolumen aus, sondern schützt den Embryo auch wie ein Kissen gegen den Druck der Gebärmutterwand der Mutter. Das Oligohydramnion führt zu einer besonderen Form des Fötus, der so genannten „Potter-Facies“, die durch Merkmale wie einen abgeflachten Nasenrücken, ein hypoplastisches Kinn, Epikanthusfalten, Katarakt und tief angesetzte Ohren gekennzeichnet ist.
Die Hauptursache des Potter-Syndroms ist unbekannt; dieses Syndrom hat in einigen Fällen einen genetischen Hintergrund und tritt häufiger bei Neugeborenen mit einer Familiengeschichte von Nierenanomalien auf. Das Syndrom, das bei 1 von 2.000 bis 5.000 Föten auftritt, ist mit einem Wiederholungsrisiko von 3 bis 6 % verbunden und wird bei 0,2 bis 0,4 % der Autopsien von toten oder unmittelbar nach der Geburt verstorbenen Neugeborenen festgestellt. Es wird jedoch vermutet, dass die Prävalenz der Krankheit höher ist, weil die betroffenen Föten tot geboren werden oder kurz nach der Geburt sterben. Es gibt keine bekannte Methode zur Vorbeugung dieser tödlichen Krankheit. Daher wird bei Müttern mit einer positiven Schwangerschaftsanamnese für dieses Syndrom ein Screening-Ultraschall in der 16. bis 18. Schwangerschaftswoche empfohlen, um Oligohydramnion und fetale Nieren zu untersuchen. Obwohl dieses Syndrom tödliche Folgen hat und nicht mit dem Leben vereinbar ist und mit dem Tod des Babys endet, erfordern die betroffenen Föten eine Wiederbelebung und eine Behandlung der Harnausgangsobstruktion zum Zeitpunkt der Entbindung. Ziel der vorliegenden Studie war es, über einen Fötus mit Potter-Syndrom zu berichten, der durch eine normale Entbindung geboren wurde.
Fallbericht
Bei dem Fall handelte es sich um einen 25 Wochen alten männlichen Fötus mit Potter-Syndrom, der durch eine normale Entbindung geboren wurde. Die Mutter wurde am 10. Mai 2017 in die Entbindungsstation des Amir al-Momenin-Krankenhauses in Zabol (Iran) eingeliefert, wo eine Ultraschalluntersuchung das Fehlen von Fruchtwasser und die Diagnose Potter-Syndrom für den Fötus ergab. Die Mutter war 24 Jahre alt, Iranerin, Hausfrau, gravid 3 und para 1 und hatte eine Abtreibungsanamnese im ersten Monat. Sie erinnerte sich nicht an den ersten Tag ihrer letzten Menstruation, und ihr Schwangerschaftsalter betrug laut dem einzigen Ultraschall, der am selben Tag durchgeführt wurde, 25 Wochen. Sie hatte eine normale Geburtsgeschichte (ihr vorheriges Kind war gesund und hatte eine normale Geburt) und war während der Schwangerschaft pränatal betreut worden. Ihre Schwangerschaftstests waren normal. In der Anamnese gab es keine Hinweise auf Rauchen, Drogen oder Alkohol, Grunderkrankungen wie Diabetes, Bluthochdruck, Schilddrüsen-, Herz-Lungen- und Nierenerkrankungen, Infektionen und Medikamenteneinnahme außer der routinemäßigen Einnahme von Schwangerschaftsmedikamenten (Eisen- und Vitaminpräparate). Die Mutter und ihr Ehemann waren Cousins und Cousinen, und sie erwähnte keine familiäre Vorgeschichte einer Neugeborenengeburt mit einer ähnlichen Störung. Pränatale serologische Tests waren negativ für Syphilis, AIDS, Hepatitis B und Röteln. Sie hatte die Blutgruppe B positiv.
Nach der erwähnten Ultraschalluntersuchung betrug das Gestationsalter 25 Wochen, der Fruchtwasserindex war Null, die Nieren des Fötus waren in der normalen anatomischen Position nicht sichtbar, die Blase war nicht sichtbar, und es gab eine schwere fetale Wachstumsrestriktion und eine schwere Kardiomegalie (mehr als 80 %). The diagnosis was Potter syndrome based on ultrasound, after which the mother was taken to the maternity ward.
The mother was hospitalized and, after the normal course of labor, gave birth to a male new-born through vaginal method. The boy had growth limitation (800 g birth weight), the clinical presentation of Potter syndrome, and severe respiratory distress and died immediately after delivery (Figures 1 and 2).
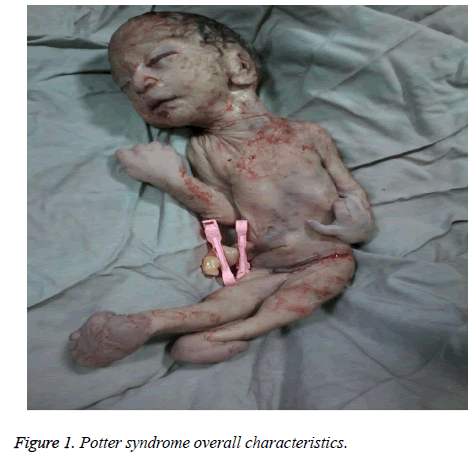
Figure 1: Potter syndrome overall characteristics.
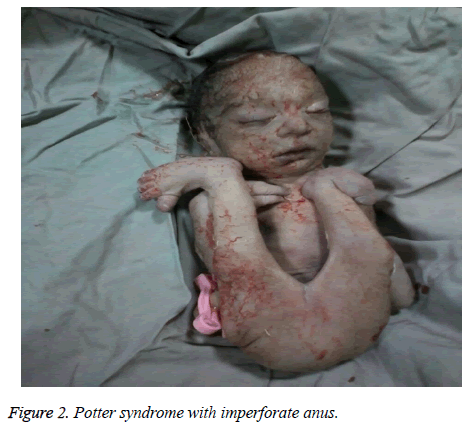
Figure 2: Potter syndrome with imperforate anus.
Discussion
The present study introduced a rare case that has not been reported in the region so far. It was a fatal case of Potter syndrome that was born through normal delivery. Bei der Ultraschalluntersuchung der Frau wurde in der 25. Schwangerschaftswoche ein Fötus mit Potter-Syndrom und einem Fruchtwasserindex von Null festgestellt. Die Mutter wurde in die Entbindungsstation eingeliefert, und das Kind wurde vaginal geboren. Das Neugeborene mit dem klinischen Bild des Potter-Syndroms litt unter schwerer Atemnot und starb kurz nach der Geburt.
Das Potter-Syndrom, das erstmals von Edith Potter bei Neugeborenen beschrieben wurde, ist durch eine bilaterale Nierenagenesie oder andere Nierenanomalien wie Aplasie, Dysplasie, Hypoplasie oder polyzystische Nierenerkrankung gekennzeichnet. Es wird eher bei Jungen erkannt, wobei das Verhältnis von Männern zu Frauen 2 zu 1 beträgt, was auf das Vorhandensein bestimmter Gene auf dem Chromosom Y hindeutet. Das Potter-Syndrom kann bei Neugeborenen mit normalen Nieren auftreten, aber die Mutter hatte in den mittleren Schwangerschaftswochen einen chronischen und anhaltenden Fruchtwasseraustritt.
In dieser Studie ergab die Ultraschalluntersuchung der Schwangeren einen Fall von Potter-Syndrom bei einem männlichen Fötus in der 25. Die Ultraschallbefunde von Föten mit schweren Nierenerkrankungen in 23 Familien wiesen auf ein verlängertes Oligohydramnion, eine schwere Nierenfunktionsstörung und das Potter-Syndrom hin, und die Säuglinge starben innerhalb von Stunden bis Tagen nach der Geburt. Moreira und Reuvers wiesen auch auf die Geburt von Föten mit Potter-Syndrom hin.
Zu den neonatalen Merkmalen des Potter-Syndroms gehören Gesichtsveränderungen, Fehlbildungen der Gliedmaßen, fetale Wachstumseinschränkung und pulmonale Hypoplasie, bekannt als Oligohydramnionstetrade. Diese Merkmale ergeben sich aus der fetalen Kompression aufgrund des verlängerten Oligohydramnionsstadiums. Das Potter-Gesicht ist gekennzeichnet durch Hypertelorismus, mongolisches Augenlid, Epikanthusfalte, abgeflachten Nasenrücken und Ohrläppchen, Papageienschnabelnase, tief angesetzte Ohren, vertieftes Kinn, eine kleine Falte unterhalb der Lippe, kurzen Hals und zusätzliche Ketten um den Hals. Das klinische Erscheinungsbild des Säuglings in dieser Studie war ein vertieftes Gesicht, ein abgeflachter Nasenrücken und abgeflachte Ohrläppchen, eine Papageienschnabelnase, tief angesetzte Ohren, ein kleines, kurzes und vertieftes Kinn, eine kleine Falte unter der Lippe, ein kurzer Hals und zusätzliche Falten um den Hals, breite Hände, eine Handabweichung am Handgelenk, ein bilateraler Valgus und eine schwere fetale Wachstumseinschränkung, die zu einem Geburtsgewicht von 800 g führte. Diese klinischen Manifestationen stimmten mit denen von Shastry und Al-Haggar überein.
Der konstante Druck der Uteruswände auf die Brustwand des Fötus und der Druck der intraabdominalen Organe auf das Zwerchfell sind eine der Folgen des Oligohydramnions beim Potter-Syndrom, die auch die Hauptursache für die Lungenhypoplasie und das Lungenversagen bei diesem Syndrom sind. Der Schweregrad der Lungenhypoplasie hängt von der Entwicklungsphase der Lunge ab, in der das Oligohydramnion auftritt, sowie von der Intensität und Dauer des Oligohydramnion. Aufgrund der schweren Atemnot und der Lungenhypoplasie werden die Föten mit dysplastischen Nieren geboren oder sterben kurz nach der Geburt. In dieser Studie lag der Fruchtwasserindex laut Ultraschall bei Null, und das Neugeborene litt bei der Geburt unter schwerer Atemnot und starb wenige Augenblicke nach der Geburt.
Das Potter-Syndrom kann mit angeborenen Herzanomalien, gastrointestinalen Anomalien (wie Ösophagusatresie, Kolonagenesie und Anal- und Duodenalfehlbildungen, Meckel-Divertikel sowie Pankreas- und Milzzysten), Skelettstörungen, Hirnanomalien und verschiedenen Assoziationen einhergehen, wie Vakarl et al. In der vorliegenden Studie wurde bei der Ultraschalluntersuchung eine schwere Kardiomegalie (mehr als 80 %) und eine unsichtbare Blase festgestellt, und bei der körperlichen Untersuchung des Neugeborenen wurden Anomalien der Gliedmaßen und ein imperforierter Anus festgestellt.
Die Hauptursache des Potter-Syndroms bleibt in den meisten Fällen unklar, aber in einigen Fällen hat es eine genetische Ursache, und das Vererbungsmuster hängt von einer bestimmten genetischen Ursache ab. Genetische Anomalien wie die autosomal dominante oder rezessive Vererbung der polyzystischen Nierenerkrankung, die hereditäre Nierendysplasie, die durch RET- und UPK3A-Genmutationen und Chromosomenanomalien verursacht wird, können zu Entwicklungsanomalien führen und das Potter-Syndrom hervorrufen. Dieses Syndrom tritt sporadisch auf, kann aber vererbt werden, wenn es aus der autosomal-dominanten Triade hervorgeht. Das Potter-Syndrom tritt häufiger bei Säuglingen auf, in deren Familie Anomalien der Nieren aufgetreten sind. In der vorliegenden Studie wurde keine spezifische Ursache für diese Krankheit gefunden, und es gab keine Familienanamnese von Nierenanomalien. San und Samal wiesen in ihren Studien auch auf die unbekannte Ätiologie des Potter-Syndroms und seine genetischen Ursachen hin.
Obwohl die Hauptursache des Potter-Syndroms die Anomalie in der Entwicklung der Nieren ist, wird die Erstdiagnose durch Ultraschall durchgeführt, der den Verlust oder das Fehlen von Fruchtwasser und das Fehlen der Blase zeigen kann, und wird durch die Untersuchung des Vorhandenseins oder Fehlens von Nieren fortgesetzt. Eine genetische Beratung ist wichtig, um die Diagnose zu bestätigen. Die Identifizierung dieses Syndroms, das durch beidseitige Nierenagenesie, Lungenhypoplasie, Potter-Fazies und Fehlbildungen der Gliedmaßen gekennzeichnet ist, kann aufgrund des Oligohydramnios per Ultraschall problematisch sein. In dieser Studie zeigte der Ultraschall das Fehlen des Fruchtwasserindexes, die Unsichtbarkeit der Blase und die beidseitige Nierenagenesie.
Das Potter-Syndrom ist mit dem Leben unvereinbar und hat eine tödliche Prognose. Die Schwangerschaft kann abgebrochen werden, bevor der Fötus das Lebensstadium erreicht. Die Standard-Schwangerschaftsbetreuung ändert sich nicht, wenn beschlossen wird, die Schwangerschaft fortzusetzen. Eine der Einschränkungen der vorliegenden Studie bestand darin, dass keine Gentests durchgeführt wurden, da die Eltern eine Autopsie ablehnten.
In der vorliegenden Studie wurde eine Mutter in der 25. Schwangerschaftswoche mit einem durch Ultraschall diagnostizierten Potter-Syndrom-Fötus in die Entbindungsstation eingeliefert und brachte ein Baby zur Welt, das wenige Augenblicke nach der Geburt starb.
Schlussfolgerung
Das Potter-Syndrom ist eine sehr ernste Krankheit und ein tödlicher Zustand. Pränataler Ultraschall hilft bei der Erkennung dieses Syndroms durch die Untersuchung von Oligohydramnion und Nierenerkrankungen.
Interessenkonflikt
Die Autoren erklären, dass sie keinen Interessenkonflikt bezüglich der Veröffentlichung dieses Fallberichts haben.
Finanzierung
Im Zusammenhang mit diesem Fallbericht wurde keine Finanzierung beantragt oder gesichert.
Patienteneinwilligung
Erhalten
Provenienz und Peer Review
Dieser Fallbericht wurde von einem Peer Reviewer geprüft.
- Potter EL. Gesichtscharakteristika von Säuglingen mit beidseitiger Nierenagenesie. Am J Obstet Gynecol 1946; 51: 885-888.
- Srihari Babu M, Asha Latha D, Radhika P. Potters syndrome: A case report. JDMS 2015; 14: 14-16.
- Sudhanshu Kumar D, Sidharth Sankar M. Potter facies with polycystic kidney disease in association with other congenital anomalies: two case reports. J Med Dent Sci 2013; 2: 2665-2668.
- Khatami F. Potters syndrome: A study of 15 patients. Arch Iranian Med 2004; 7: 186- 189.
- Sarkar S, Das Gupta S, Barua M, Ghosh R, Mondal K, Chatterjee U, Datta C. Potters sequence: Eine Geschichte des Seltenen, Seltenen und des Seltensten. Indian J Pathol Microbiol 2015; 58: 102-104.
- Manoj MG, Kakkar S. Potters syndrome-A fatal constellation of anomalies. Indian J Med Res 2014; 139: 648-649.
- Himabindu A, Narasinga B. A fatal case of potters syndrome-a case report. JCDR 2011; 5: 1264-1266.
- Moreira BL, Monarim MA, Romano RF, Mattos LA, DIppolito G. Doege-Potter syndrome. Radiol Bras 2015; 48: 195-196.
- Reuvers JR, van Dorp M, Van Schil PE. Solitärer fibröser Tumor der Pleura mit assoziiertem Doege-Potter-Syndrom. Acta Chir Belg 2016; 116: 386-387.
- Shastry SM, Kolte SS, Sanagapati PR. Potters sequence. J Clin Neonatol 2012; 1: 157-159.
- Al-Haggar M, Yahia S, Abdel-Hadi D, Grill F, Al-Kaissi A. Sirenomelie (Symelia apus) mit Potters-Syndrom in Verbindung mit Gestationsdiabetes mellitus: Ein Fallbericht und Literaturübersicht. Afr Health Sci 2010; 10: 395-399.
- Dhundiraj KM, Madhukar DN, Ambadasrao PG, Wamanrao KS, Prem ZM. Potters Syndrom: A report of 5 cases. Indian J Pathol Microbiol 2006; 49: 254-257.
- Dayal J, Maheshwari E, Ghai PS, Bhatotia S. Antenatal ultrasound diagnosis of Potters syndrome. Indian J Radiol Imaging 2003; 13: 81-83.
- San Agustin JT, Klena N, Granath K, Panigrahy A, Stewart E, Devine W, Strittmatter L, Jonassen JA, Liu X. Genetic link between renal birth defects and congenital heart disease. Nat Commun 2016; 22: 111-118.
- Samal SK, Rathod S. Sirenomelia: The mermaid syndrome: report of two cases. J Nat Sci Biol Med 2015; 6: 264-266.
- Gerards FA, Twisk JW, Fetter WP. Vorhersage der pulmonalen Hypoplasie mit 2- oder 3-dimensionaler Sonographie bei komplizierten Schwangerschaften. Am J Obstet Gynecol 2008; 198: 140-146.
- Kemper MJ, Mueller-Wiefel DE. Prognosis of antenatally diagnosed oligohydramnios of renal origin. Eur J Pediatr 2007; 166: 393-398.