Einführung
Das kaudale Regressionssyndrom (CRS) ist eine seltene Störung, die erstmals 1852 von Geoffroy Saint-Hilaire und Hohl beschrieben wurde. 1964 prägte Duhmel den Begriff „kaudales Regressionssyndrom“.1-3 Das CRS ist eine Störung, die durch eine Anomalie der distalen Wirbelsäulensegmente verursacht wird, und erstreckt sich auf ein breites Spektrum von Anomalien wie partielle Agenesie des Rückenmarks, damit verbundene Beckenfehlbildungen, imperforierter Anus, genitale Fehlbildungen, Herzanomalien, bilaterale Nierendysplasie oder -aplasie, pulmonale Hypoplasie und extreme Außenrotation mit Fusion der inferioren Gelenke, die in der schwersten Form in Sirenomelie (Meerjungfrau-Syndrom) resultiert. Das CRS geht auch mit einer Femurhypoplasie, deformierten Füßen und einer Flexionskontraktur der unteren Extremitäten einher. Die Intelligenz bleibt im Allgemeinen erhalten.1,2,4 Es betrifft zwischen 0,1 und 0,25 von 10.000 Schwangerschaften, wobei das Verhältnis zwischen Männern und Frauen 2,7:1 beträgt.3-5 Auf embryonaler Ebene geht man davon aus, dass das CRS das Ergebnis von Defekten bei der Induktion von kaudalen Elementen im Embryo vor dem 28. Die Verletzung entsteht in der hinteren medialen mesodermischen Achse und führt zum Fehlen der Entwicklung des kaudalen mesoblastischen Dottersaftes.4,6 Die genaue Ätiologie ist unbekannt, jedoch wurden mütterlicher Diabetes, genetische Prädisposition und vaskuläre Hypoperfusion als mögliche Faktoren vorgeschlagen.4-7
Der Schwangerschaftsdiabetes ist zweifellos ein Teratogen, und es gibt gute Hinweise darauf, dass der Schwangerschaftsdiabetes an der Entwicklung der schwersten Form des CRS beteiligt ist.5
Pinter und Reece haben nachgewiesen, dass zu den durch Hyperglykämie verursachten Veränderungen beim Abschluss des Neuralrohrs ungeordnete Zellen, ein Rückgang der Mitose und Veränderungen gehören, die auf eine vorzeitige Reifung hinweisen. Der durch den mütterlichen Diabetes veränderte oxidative Stoffwechsel kann zu einer erhöhten Produktion von sauerstofffreien Radikalen im sich entwickelnden Embryo führen, die teratogen sein können.8 Zwischen 16 % und 22 % der CRS-Fälle sind mit mütterlichem Diabetes mellitus assoziiert, was das Risiko, ein Kind mit CRS zu bekommen, um bis zu 400 erhöht.3,4,7 Es wurden mehrere Fälle von Familien mit CRS gemeldet, was auf eine mögliche genetische Übertragung mit verschiedenen möglichen Übertragungsmodi schließen lässt: X-chromosomal-dominante, multifaktorielle polygene und autosomal-dominante Muster mit reduzierter Penetranz und variabler Expressivität.7 Die Theorie des „vaskulären Diebstahls“ wurde ursprünglich 1927 von Kampmeier vorgeschlagen und 1986 von Stevenson wieder aufgegriffen. Adra et al. betrachteten Stevensons „vaskulärer Diebstahl“-Theorie als mögliche Ätiologie der CRS-Pathologie.7 Während der Embryonalentwicklung werden die eher kaudalen Strukturen von den kephalen Elementen wie dem Gehirn, der Wirbelsäule und dem Rückenmark getrennt, daher das Fehlen von kognitiven Veränderungen bei diesem Syndrom.4
Es gibt zwei CRS-Gruppen: Die erste Gruppe ist die am stärksten betroffene mit dem Ende des Rückenmarks oberhalb von L1. Das Kreuzbein endet bei S1, und in einigen Fällen fehlt es sogar ganz. Die Patienten der zweiten Gruppe weisen eine weniger schwere Dysgenese mit einer niedrigen Implantation des Rückenmarks auf, die durch ein verdicktes Filum terminale oder ein intraspinales Lipom gefesselt ist.4
Falldarstellung
Wir stellen den Fall eines neugeborenen Mädchens einer 40-jährigen Mutter mit einer Anamnese unbehandelter Uterusmyome, einem Hintergrund unregelmäßiger gynäkologischer Zyklen, 3 Schwangerschaften mit 2 Entbindungen und einer Cholezystektomie im Jahr 2008 vor. Sie berichtet von einer ungeplanten, aber dennoch gewollten Schwangerschaft, bei der eine Risikoschwangerschaftsvorsorge durchgeführt wurde. Sie nahm an über 15 pränatalen Beratungen teil. In der 18. Schwangerschaftswoche wurden kompatible Daten mit Fehlbildungen des Neuralrohrs mit Sakralagenesie gefunden, was zur Diagnose CRS führte. Während des zweiten Trimesters der Schwangerschaft wurde eine Hyperglykämie festgestellt, weshalb sie mit Insulin behandelt wurde. In der 40. Schwangerschaftswoche wurde ein Kaiserschnitt durchgeführt, bei dem ein gesundes Kind mit einem Apgar-Score von 8/9 und einem Gestationsalter von 39,6 Wochen zur Welt kam. Bei der körperlichen Untersuchung stellten wir ein Gewicht von 3,44 kg, eine Länge von 49 cm, einen Kopfumfang von 35 cm und einen Bauchumfang von 32 cm fest. Bei der Inspektion konnten wir eine offensichtliche diabetische Fetopathie mit reichlich Haaren und geringer Implantation feststellen (Abb. 1). Normale vordere Fontanelle (2 x 2 cm), rundes Gesicht, ausgeprägte Wangenknochen, horizontale Lidspalten, kurzer Nasenrücken, runde Spitze, dünne Lippen, voller Gaumen, Dysplasie der Ohrmuscheln mit Hypertrichose der Helix, Kurzer Hals mit dorsaler Ausbuchtung, normaler Thorax ohne Auskultationsgeräusche, weicher Bauch ohne tastbare Massen oder Visceromegalie, geradlinige Wirbelsäule mit einem Grübchen auf der Haut im Lumbosakralbereich (Abb. 2). 2), kein Kreuzbeinknochen war tastbar, der Anus lag normal, war durchlässig und hatte keinen Tonus. Es gab eine deutliche Verkürzung der unteren Extremitäten und einen beidseitigen Varus-Klumpfuß (Abb. 3). Es wurden Röntgenaufnahmen angefertigt, um die Knochenfehlbildungen zu sehen, und wir konnten eine vollständige Agenesie des Kreuzbeins mit einer lumbal-iliakalen Fusion feststellen. Eine Magnetresonanztomographie (MRT) des Gehirns und der Wirbelsäule (Abb. 4) zeigte ein abruptes Ende der Sehne und der Lendenwirbelsäule auf Höhe von L3. Hinter dieser Ebene konnten wir nur amorphe Massen mit Fettsignalintensität erkennen, die mit subkutanem Gewebe in Verbindung stehen. Kurzes Rückenmark, flacher Conus medullaris auf Höhe von T10, beide Beckenknochen waren hypoplastisch und auf halber Höhe verschmolzen. Das Kreuzbein ist nicht zu sehen. Es war möglich, beide Nieren malrotiert und hypoplastisch zu sehen, eine prominente Harnblase, die mit einer neurogenen Blase vereinbar ist. Das Gehirnbild wurde als normal bezeichnet. Es wurde eine Oberbauchechographie angefordert, um andere viszerale Anomalien auszuschließen. Dabei wurde eine diskrete Diskrepanz in der Größe der linken Niere festgestellt, die zudem nicht normal geformt war, was auf eine diskrete Hypoplasie und eine mögliche Malrotation schließen ließ. Während ihres Aufenthalts führten wir eine multidisziplinäre Beratung durch, und beide unteren Gliedmaßen wurden mit Gipsschienen versorgt. Die Patientin wird weiterhin von der Urologie, der Traumatologie, der Neurologie und der allgemeinen Pädiatrie betreut, um alle Risikofaktoren und möglichen Komplikationen genau im Auge zu behalten.
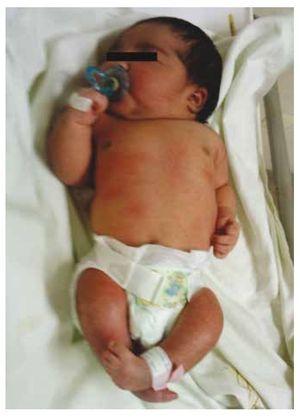
Abbildung 1 Neugeborenes mit diabetischer Fetopathie.
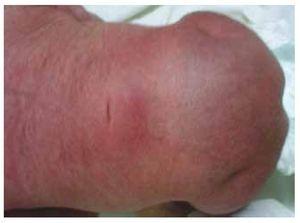
Abbildung 2 Ein Grübchen auf der Haut in der Lumbosakralregion.
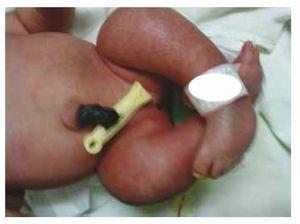
Figure 3 Equinovarus feet.
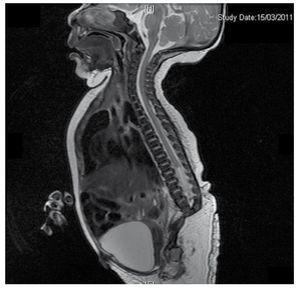
Figure 4 The lumbar spine magnetic resonance imaging (MRI) shows an abrupt chord and lumbar termination at the L3 level. Short spinal cord, flat conus medullaris at T10 level, hypoplastic iliac bones, fused at mid-level. No sacrum is seen.
Discussion
The CRS is a rare congenital malformation. Even though the specific etiologic factor is unknown, it is related to maternal diabetes, genetic predisposition and vascular hypoperfusion.3 Just as in the case presented, this alteration is characterized by agenesis of the sacrum involving iliac and lumbar vertebrae with their corresponding spinal segments and variable abnormalities in the lower limbs as well as in other organs. Fetal diagnosis tools allow early syndrome detection. Der pränatale Ultraschall ist das am häufigsten verwendete paraklinische Instrument; ein Schlüsselelement des pränatalen Ultraschalls ist die detaillierte Beurteilung der Wirbelsäule und der unteren Gliedmaßen; er ermöglicht auch eine CRS-Diagnose durch den Nachweis eines abrupten Endes der Lendenwirbelsäule und hypoplastischer unterer Gliedmaßen. Bei der pränatalen Diagnose müssen wir uns darauf konzentrieren, den Grad der Digenesie sowie die damit verbundenen kongenitalen Anomalien zu erkennen, um eine Prognose und einen rechtzeitigen Plan für postnatale therapeutische Interventionen aufzustellen.3,5 Die 1978 geschaffene Klassifikation von Renshav teilt das Syndrom in 4 Grade ein, die sich nach dem Schweregrad der Kreuzbeinagenesie und der Beteiligung von Darmbein- und Lendenwirbeln richten.9 Nach dieser Klassifikation gehört unsere Patientin zum Grad iV (vollständige Kreuzbeinagenesie mit Verschmelzung der Darmbeine). Diese Gruppe ist mit einer noch schlechteren Prognose verbunden, mit größeren neurologischen Auswirkungen und multisystemischen Folgeerscheinungen, vor allem auf Nierenebene. Dieser Fall ist ein deutliches Beispiel für das breite Spektrum an Veränderungen, die den heranwachsenden Fötus als Folge eines unkontrollierten mütterlichen Diabetes während der Schwangerschaft beeinträchtigen können. Aufgrund der hohen Korrelation zwischen diesem Defekt und der diabetischen Mutter und seiner Entwicklung in den frühen Stadien der Schwangerschaft ist eine Präventionsstrategie, die eine strenge Blutzuckerkontrolle vor der embryonalen Organogenese oder bei Hochrisikopatientinnen sogar noch früher vorsieht, unerlässlich. Eine gute Beratung und genetische Untersuchungen vor der Schwangerschaft sind ebenfalls wichtig. Die Behandlung ist sowohl für den Arzt als auch für die Eltern eine Herausforderung und erfordert einen multidisziplinären Ansatz, an dem je nach Schweregrad ein Kinderarzt, ein Kinderchirurg, ein Orthopäde, ein Physiotherapeut und ein Urologe beteiligt sind. Given the fact that the primary pathology is irreversible, treatment is just supportive, with the sole purpose of accomplishing a life as normal as possible.
Conflicts of interest
The authors have no conflicts of interest to declare.
Funding
No financial support was provided.
Received: September 2013;
Accepted: January 2014