Informe de un caso – Investigación Biomédica (2019) Volumen 30, Número 2
Informe de un caso mortal de síndrome de potter: Un informe de caso.
Mehrbano Amirshahi, Mahin Badakhsh* y Zohreh Sadat Hashemi
Departamento de Partería, Escuela de Enfermería y Partería, Universidad de Ciencias Médicas de Zabol, Irán
*Autor correspondiente: Mahin Badakhsh
Departamento de Partería
Escuela de Enfermería y Partería
Universidad de Ciencias Médicas de Zabol
Irán
Fecha de aceptación: 21 de febrero de 2019
DOI: 10.35841/biomedicalresearch.30-19-084
Visita para ver más artículos relacionados en Biomedical Research
Abstract
Antecedentes: El síndrome de Potter es un trastorno congénito poco frecuente que hace referencia a un conjunto de manifestaciones clínicas que se asocian a un oligohidramnios causado por una insuficiencia renal fetal. El rasgo distintivo de este síndrome es el especial cuadro clínico que además de oligohidramnios se caracteriza por hipoplasia pulmonar, agenesia renal bilateral; deformidades de las extremidades y cara específica. El embrión muere antes o inmediatamente después del nacimiento por insuficiencia respiratoria. El objetivo de este estudio es informar de un feto con síndrome de Potter que nació por parto vaginal. Informe del caso: El examen ecográfico de la mujer embarazada reveló que su feto masculino con edad gestacional de 25 semanas con síndrome de Potter y el índice de líquido amniótico es cero. La madre fue hospitalizada en la unidad de parto y dio a luz de forma natural. El bebé presentaba un cuadro clínico de síndrome de Potter y dificultad respiratoria grave y murió poco después del nacimiento. Conclusiones: El síndrome de Potter es una enfermedad muy grave y la mayoría de las veces es mortal. La ecografía prenatal mediante el examen del oligohidramnios y los riñones ayuda al diagnóstico.
Palabras clave
Síndrome de Potter, Oligohidramnios, Hipoplasia pulmonar, Feto
Introducción
El síndrome de Potter es una rara malformación congénita que afecta principalmente a los fetos masculinos y se caracteriza por una hipoplasia pulmonar causada por un fallo renal. Fue notificado por primera vez por Edith Potter en 1946. Después de la semana 16 de embarazo, la cantidad de líquido amniótico depende principalmente de la producción de orina del feto. Durante la vida intrauterina, el feto traga continuamente el líquido amniótico, que vuelve al saco amniótico por los riñones. El oligohidramnios se produce cuando el volumen de líquido amniótico es inferior al normal para ese periodo del embarazo. Esta disminución del volumen puede deberse a una reducción de la producción de orina, secundaria a causas como la agenesia renal bilateral, la obstrucción de las vías urinarias y la rotura prolongada de las bolsas amnióticas. La orina del feto, esencial para el desarrollo de los pulmones, desempeña su papel contribuyendo al desarrollo de las vías respiratorias y los alvéolos, creando presión hidráulica y proporcionando prolina, un aminoácido esencial para el desarrollo de los pulmones. Si los alvéolos y, por lo tanto, los pulmones no se desarrollan adecuadamente al nacer, el neonato no podrá respirar bien y sufrirá de dificultad respiratoria debido a la hipoplasia pulmonar. Por lo tanto, secundaria a la insuficiencia renal, la hipoplasia pulmonar es la principal causa de muerte en los neonatos con síndrome de Potter.
La producción de orina por parte del embrión no sólo afecta al volumen de líquido amniótico, sino que también preserva al embrión frente a la presión de la pared del útero de la madre como un cojín. El oligohidramnios da lugar a una forma especial de feto denominada «facies de Potter», que se caracteriza por rasgos como puente nasal aplanado, mentón hipoplásico, pliegues epicánticos, cataratas y orejas de implantación baja.
La causa principal del síndrome de Potter es desconocida; este síndrome tiene un trasfondo genético en algunos casos, y es más frecuente en neonatos con antecedentes familiares de anomalías renales . El síndrome, con una incidencia de 1 de cada 2.000 a 5.000 fetos, se asocia a un riesgo de recurrencia del 3-6%, y se encuentra en el 0,2-0,4% de las autopsias de recién nacidos muertos o que fallecen inmediatamente después del nacimiento . Sin embargo, se cree que la enfermedad puede tener una mayor prevalencia porque los fetos afectados nacen muertos o mueren poco después del parto. No se conoce ningún método para prevenir esta enfermedad mortal. Por lo tanto, se recomienda realizar una ecografía de cribado a las 16-18 semanas de gestación a las madres con un historial positivo de embarazo para este síndrome con el fin de evaluar el oligohidramnios y los riñones del feto. Aunque este síndrome tiene consecuencias mortales y no es compatible con la vida y termina con la muerte del bebé, los fetos afectados requieren reanimación y tratamiento de la obstrucción de la salida urinaria en el momento del parto . El presente estudio tiene como objetivo informar de un feto con síndrome de Potter que nació a través de un parto normal.
Informe del caso
El caso era un feto masculino de 25 semanas con síndrome de Potter, nacido a través de un parto normal. La madre ingresó en la maternidad del Hospital Amir al-Momenin, en Zabol, Irán, el 10 de mayo de 2017 con una ecografía, que mostró la ausencia de líquido amniótico y el diagnóstico de síndrome de Potter para el feto. La madre tenía 24 años, era iraní, ama de casa, grávida 3 y para 1, y tenía antecedentes de aborto en el mes 1. No recordaba el primer día de su última menstruación, y su edad gestacional era de 25 semanas según la única ecografía realizada el mismo día. Tenía un historial de parto normal (su hijo anterior estaba sano y tuvo un parto normal), y había recibido atención prenatal durante el embarazo. Sus pruebas de embarazo eran normales. No había antecedentes de tabaquismo, drogas o alcohol, enfermedades subyacentes como diabetes, hipertensión, tiroides, cardiopulmonares y renales, infecciones, ni consumo de fármacos aparte de los medicamentos habituales para el embarazo (suplementos de hierro y vitaminas). La madre y su marido eran primos y ella no mencionó antecedentes familiares de un parto neonatal con un trastorno similar. Las pruebas serológicas prenatales fueron negativas para la sífilis, el SIDA, la hepatitis B y la rubéola. Tenía un grupo sanguíneo B positivo.
Según la ecografía mencionada, la edad gestacional era de 25 semanas, el índice de líquido amniótico era cero, los riñones del feto eran invisibles en la posición anatómica normal, la vejiga era invisible, y había una severa restricción del crecimiento fetal y una cardiomegalia severa (más del 80%). The diagnosis was Potter syndrome based on ultrasound, after which the mother was taken to the maternity ward.
The mother was hospitalized and, after the normal course of labor, gave birth to a male new-born through vaginal method. The boy had growth limitation (800 g birth weight), the clinical presentation of Potter syndrome, and severe respiratory distress and died immediately after delivery (Figures 1 and 2).
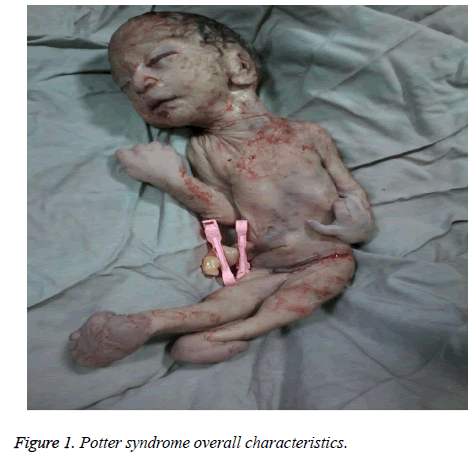
Figure 1: Potter syndrome overall characteristics.
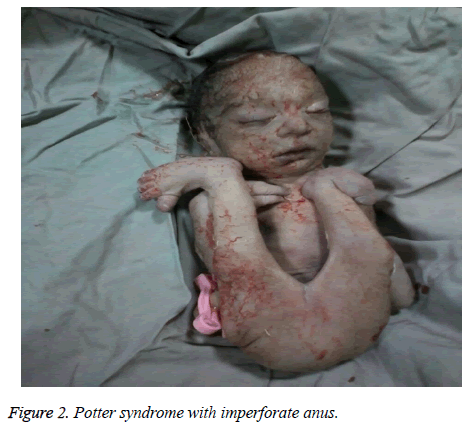
Figure 2: Potter syndrome with imperforate anus.
Discussion
The present study introduced a rare case that has not been reported in the region so far. It was a fatal case of Potter syndrome that was born through normal delivery. La ecografía de la mujer reveló un feto de 25 semanas de embarazo con síndrome de Potter y un índice de líquido amniótico de cero. La madre fue ingresada en la unidad de partos y el bebé nació por vía vaginal. El recién nacido con la presentación clínica del síndrome de Potter tenía dificultad respiratoria grave y murió poco después del nacimiento.
El síndrome de Potter, descrito por primera vez por Edith Potter en neonatos, se caracteriza por agenesia renal bilateral u otras anomalías renales como aplasia, displasia, hipoplasia o poliquistosis renal. Se reconoce más en los varones, con una proporción entre hombres y mujeres de 2 a 1, lo que sugiere la presencia de ciertos genes en el cromosoma Y . El síndrome de Potter puede observarse en neonatos con riñones normales, pero la madre tuvo una fuga crónica y prolongada de líquido amniótico durante las semanas centrales del embarazo.
En este estudio, la ecografía de las embarazadas reveló un caso de síndrome de Potter en un feto varón en la semana 25 de embarazo, un índice de líquido amniótico nulo y la ausencia de ambos riñones y de la vejiga. Los hallazgos ecográficos de los fetos con enfermedad renal grave en 23 familias indicaban oligohidramnios prolongado, disfunción renal grave y facies de Potter, y los bebés murieron entre horas y días después del nacimiento. Moreira y Reuvers también señalaron el nacimiento de fetos con síndrome de Potter.
Las características neonatales del síndrome de Potter incluyen alteraciones faciales, malformaciones de las extremidades, limitación del crecimiento fetal e hipoplasia pulmonar, conocida como tétrada de oligohidramnios. Estos rasgos surgen de la compresión fetal debida al oligohidramnios prolongado. La facies de Potter se caracteriza por hipertelorismo, párpado mongólico, pliegue epicántico, puente nasal y lóbulo de la oreja aplanados, nariz de pico de loro, orejas de implantación baja, mentón hundido, un pequeño pliegue debajo del labio, cuello corto y cadenas adicionales alrededor del cuello. La presentación clínica del bebé de este estudio era cara hundida, puente nasal y lóbulo de la oreja aplanados, nariz de pico de loro, orejas de implantación baja, barbilla pequeña, corta y hundida, un pequeño pliegue debajo del labio, cuello corto y pliegues adicionales alrededor del cuello, manos anchas, desviación de la mano en la muñeca, valgo bilateral y limitación grave del crecimiento fetal, lo que dio lugar a un peso al nacer de 800 g. Estas manifestaciones clínicas coincidían con las de Shastry y Al-Haggar.
La presión constante de las paredes uterinas sobre la pared torácica del feto y la presión de los órganos intraabdominales sobre el diafragma constituyen una de las consecuencias del oligohidramnios en el síndrome de Potter, que son también las principales causas de la hipoplasia y el fallo pulmonar en este síndrome. La gravedad de la hipoplasia pulmonar depende de la fase de desarrollo del pulmón en la que se produce el oligohidramnios, así como de la intensidad y duración del mismo. Debido a la dificultad respiratoria grave y a la hipoplasia pulmonar, los fetos nacen con riñones displásicos o mueren poco después del nacimiento. En este estudio, el índice de líquido amniótico era nulo según la ecografía y el recién nacido presentaba una grave dificultad respiratoria al nacer y murió a los pocos momentos de nacer.
El síndrome de Potter puede asociarse a anomalías cardíacas congénitas, anomalías gastrointestinales (como atresia de esófago, agenesia de colon y malformaciones anales y duodenales, divertículo de Meckel y quistes pancreáticos y de bazo), trastornos esqueléticos, anomalías cerebrales y diversas asociaciones, como Vakarl et al. . En el presente estudio, la ecografía informó de una cardiomegalia grave (más del 80%) y una vejiga invisible; y el examen físico del neonato reveló anomalías en las extremidades y un ano imperforado.
La causa principal del síndrome de Potter sigue sin estar clara en la mayoría de los casos, pero tiene una razón genética en algunos casos, y el patrón de herencia depende de una causa genética concreta. Las anomalías genéticas, como la herencia autosómica dominante o recesiva de la poliquistosis renal, la displasia renal hereditaria, causada por mutaciones de los genes RET y UPK3A y las anomalías cromosómicas, pueden dar lugar a anomalías del desarrollo y provocar el síndrome de Potter. Este síndrome se produce de forma esporádica, pero puede heredarse cuando surge de la tríada autosómica dominante. El síndrome de Potter es más frecuente en bebés con antecedentes familiares de anomalías renales. En el presente estudio no se encontró ninguna causa específica para esta enfermedad, y no había antecedentes familiares de anomalías renales. San y Samal también señalaron la etiología desconocida del síndrome de Potter y sus razones genéticas en sus estudios.
Aunque la causa principal del síndrome de Potter es la anormalidad en el desarrollo de los riñones, el diagnóstico inicial se realiza mediante ecografía que puede mostrar la pérdida o ausencia de líquido amniótico y la ausencia de vejiga, y se continúa mediante la investigación sobre la presencia o ausencia de riñones. El asesoramiento genético es importante para confirmar el diagnóstico. La identificación de este síndrome, que se caracteriza por agenesia renal bilateral, hipoplasia pulmonar, facies de Potter y malformación de las extremidades, puede ser problemática mediante la ecografía debido al oligohidramnios . En este estudio, la ecografía mostró la ausencia de índice de líquido amniótico, invisibilidad de la vejiga y agenesia renal bilateral.
El síndrome de Potter es incompatible con la vida y tiene un pronóstico mortal. El embarazo puede interrumpirse antes de que el feto alcance la fase vital. Los cuidados estándar del embarazo no cambian cuando se decide continuar con la gestación . Una de las limitaciones del presente estudio fue que no se realizaron pruebas genéticas, ya que los padres no aceptaron la autopsia.
En el presente estudio, una madre en la semana 25 de embarazo con un feto con síndrome de Potter, diagnosticado a través de una ecografía, fue ingresada en la unidad de partos y dio a luz con normalidad a un bebé que falleció a los pocos instantes de nacer.
Conclusión
El síndrome de Potter es una enfermedad muy grave y mortal. La ecografía prenatal ayuda a detectar este síndrome examinando el oligohidramnios y las afecciones renales.
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses en relación con la publicación de este informe de caso.
Financiación
No se buscó ni se consiguió financiación en relación con este informe de caso.
Consentimiento del paciente
Obtenido
Proveniencia y revisión por pares
Este informe de caso fue revisado por pares.
- Potter EL. Características faciales de los bebés con agenesia renal bilateral. Am J Obstet Gynecol 1946; 51: 885-888.
- Srihari Babu M, Asha Latha D, Radhika P. Síndrome de Potters: Un informe de caso. JDMS 2015; 14: 14-16.
- Sudhanshu Kumar D, Sidharth Sankar M. Facies de Potter con enfermedad renal poliquística en asociación con otras anomalías congénitas: informe de dos casos. J Med Dent Sci 2013; 2: 2665-2668.
- Khatami F. Potters syndrome: Un estudio de 15 pacientes. Arch Iranian Med 2004; 7: 186- 189.
- Sarkar S, Das Gupta S, Barua M, Ghosh R, Mondal K, Chatterjee U, Datta C. Potters sequence: Una historia de lo raro, lo más raro y lo más raro. Indian J Pathol Microbiol 2015; 58: 102-104.
- Manoj MG, Kakkar S. Potters syndrome-A fatal constellation of anomalies. Indian J Med Res 2014; 139: 648-649.
- Himabindu A, Narasinga B. A fatal case of potters syndrome-a case report. JCDR 2011; 5: 1264-1266.
- Moreira BL, Monarim MA, Romano RF, Mattos LA, DIppolito G. Doege-Potter syndrome. Radiol Bras 2015; 48: 195-196.
- Reuvers JR, van Dorp M, Van Schil PE. Tumor fibroso solitario de la pleura con síndrome de Doege-Potter asociado. Acta Chir Belg 2016; 116: 386-387.
- Shastry SM, Kolte SS, Sanagapati PR. Secuencia de Potters. J Clin Neonatol 2012; 1: 157-159.
- Al-Haggar M, Yahia S, Abdel-Hadi D, Grill F, Al-Kaissi A. Sirenomelia (symelia apus) con síndrome de Potters en relación con la diabetes mellitus gestacional: Un informe de caso y revisión de la literatura. Afr Health Sci 2010; 10: 395-399.
- Dhundiraj KM, Madhukar DN, Ambadasrao PG, Wamanrao KS, Prem ZM. Síndrome de Potters: Un informe de 5 casos. Indian J Pathol Microbiol 2006; 49: 254-257.
- Dayal J, Maheshwari E, Ghai PS, Bhatotia S. Antenatal ultrasound diagnosis of Potters syndrome. Indian J Radiol Imaging 2003; 13: 81-83.
- San Agustín JT, Klena N, Granath K, Panigrahy A, Stewart E, Devine W, Strittmatter L, Jonassen JA, Liu X. Relación genética entre los defectos renales de nacimiento y las cardiopatías congénitas. Nat Commun 2016; 22: 111-118.
- Samal SK, Rathod S. Sirenomelia: El síndrome de la sirena: informe de dos casos. J Nat Sci Biol Med 2015; 6: 264-266.
- Gerards FA, Twisk JW, Fetter WP. Predicción de hipoplasia pulmonar con ultrasonografía bidimensional o tridimensional en embarazos complicados. Am J Obstet Gynecol 2008; 198: 140-146.
- Kemper MJ, Mueller-Wiefel DE. Prognosis of antenatally diagnosed oligohydramnios of renal origin. Eur J Pediatr 2007; 166: 393-398.