Case Report – Biomedical Research (2019) Volume 30, Issue 2
Report of a deadly case of potter syndrome: A case report.
Mehrbano Amirshahi, Mahin Badakhsh* and Zohreh Sadat Hashemi
Department of Midwifery, School of Nursing and Midwifery, Zabol University of Medical sciences, Zabol, Iran
*Autor prowadzący: Mahin Badakhsh
Department of Midwifery
School of Nursing and Midwifery
Zabol University of Medical sciences
Iran
Accepted date: February 21, 2019
DOI: 10.35841/biomedicalresearch.30-19-084
Visit for more related articles at Biomedical Research
Abstract
Tło: Zespół Pottera jest rzadkim zaburzeniem wrodzonym, które odnosi się do zespołu objawów klinicznych, które są związane z oligohydramnios spowodowanym niewydolnością nerek płodu. Cechą charakterystyczną tego zespołu jest szczególny obraz kliniczny, który oprócz oligohydramnios charakteryzuje się hipoplazją płucną, obustronną agenezją nerek, deformacjami kończyn i specyficzną twarzą. Zarodek umiera przed lub bezpośrednio po urodzeniu z powodu niewydolności oddechowej. Celem pracy jest opisanie przypadku płodu z zespołem Pottera, który urodził się drogą pochwową. Opis przypadku: W badaniu ultrasonograficznym ciężarnej stwierdzono, że jej płód płci męskiej o wieku ciążowym 25 tygodni z zespołem Pottera, a wskaźnik płynu owodniowego wynosi zero. Matka była hospitalizowana w oddziale porodowym i urodziła w przebiegu naturalnym. Dziecko miało obraz kliniczny zespołu Pottera oraz ciężką niewydolność oddechową i zmarło wkrótce po urodzeniu. Wnioski: Zespół Pottera jest bardzo poważnym schorzeniem i w większości przypadków jest śmiertelny. W rozpoznaniu pomocne jest prenatalne badanie ultrasonograficzne, w którym bada się oligohydramnios i nerki.
Słowa kluczowe
Zespół Pottera, Oligohydramnios, Hipoplazja płucna, Płód
Wprowadzenie
Zespół Pottera jest rzadką wadą wrodzoną, która dotyczy głównie płodów męskich i charakteryzuje się hipoplazją płucną spowodowaną niewydolnością nerek. Po raz pierwszy został opisany przez Edith Potter w 1946 roku. Po 16. tygodniu ciąży ilość płynu owodniowego zależy głównie od produkcji moczu przez płód. W trakcie życia wewnątrzmacicznego płód stale połyka płyn owodniowy, który wraca z powrotem do worka owodniowego przez nerki. Oligohydramnios występują, gdy objętość płynu owodniowego jest mniejsza niż normalnie dla tego okresu ciąży. Ten spadek objętości może być spowodowany zmniejszeniem ilości oddawanego moczu, wtórnie do takich przyczyn jak obustronne zwyrodnienie nerek, niedrożność dróg moczowych czy przedłużające się pęknięcie worka owodniowego. Mocz płodu, niezbędny dla rozwoju płuc, odgrywa swoją rolę przyczyniając się do rozwoju dróg oddechowych i pęcherzyków płucnych, tworząc ciśnienie hydrauliczne i dostarczając proliny, aminokwasu niezbędnego dla rozwoju płuc. Jeśli pęcherzyki płucne i w rezultacie płuca nie są odpowiednio rozwinięte po urodzeniu, noworodek nie będzie w stanie dobrze oddychać i będzie cierpiał na zaburzenia oddychania z powodu hipoplazji płuc. Dlatego, wtórna do niewydolności nerek, hipoplazja płuc jest główną przyczyną śmierci noworodków z zespołem Pottera .
Produkcja moczu przez zarodek nie tylko wpływa na objętość płynu owodniowego, ale także chroni zarodek przed naciskiem ściany macicy matki jak poduszka. Oligohydramnios powoduje szczególną formę płodu zwaną „Potter facies”, która charakteryzuje się cechami takimi jak spłaszczony mostek nosowy, hipoplastyczny podbródek, fałdy naskórkowe, zaćma i nisko osadzone uszy .
Główna przyczyna zespołu Pottera jest nieznana; zespół ten ma podłoże genetyczne w niektórych przypadkach i jest bardziej powszechny u noworodków z rodzinną historią nieprawidłowości nerek . Zespół ten, występujący z częstością 1 na 2000-5000 płodów, wiąże się z ryzykiem nawrotu wynoszącym 3-6% i jest stwierdzany w 0,2-0,4% przypadków autopsji martwych noworodków lub tych, które zmarły bezpośrednio po urodzeniu. Uważa się jednak, że częstość występowania choroby może być większa, ponieważ dotknięte nią płody rodzą się martwe lub umierają wkrótce po porodzie. Nie jest znana metoda zapobiegania tej śmiertelnej choroby. Dlatego zaleca się wykonywanie przesiewowych badań ultrasonograficznych w 16-18 tygodniu ciąży u matek z dodatnim wywiadem w kierunku tego zespołu w celu oceny oligohydramnios i nerek płodu. Mimo, że zespół ten ma śmiertelne konsekwencje i jest nie do pogodzenia z życiem, a kończy się śmiercią dziecka, dotknięte nim płody wymagają resuscytacji i leczenia zaburzeń odpływu moczu w czasie porodu. Celem pracy było opisanie płodu z zespołem Pottera, który urodził się w wyniku prawidłowego porodu.
Raport przypadku
Przypadek stanowił 25-tygodniowy płód płci męskiej z zespołem Pottera, urodzony w wyniku prawidłowego porodu. Matka została przyjęta na oddział położniczy szpitala Amir al-Momenin, Zabol, Iran, w dniu 10 maja 2017 r. z badaniem ultrasonograficznym, które wykazało brak płynu owodniowego i rozpoznanie zespołu Pottera dla płodu. Matka miała 24 lata, była Iranką, gospodynią domową, gravid 3, i para 1, i miała historię aborcji w miesiącu 1. Nie pamiętała pierwszego dnia swojej ostatniej miesiączki, a jej wiek ciążowy wynosił 25 tygodni zgodnie z jedynym USG wykonanym tego samego dnia. Miała normalną historię porodu (jej poprzednie dziecko było zdrowe i miało normalny poród) i była objęta opieką prenatalną podczas ciąży. Jej testy ciążowe były w normie. Nie było historii palenia, narkotyków lub alkoholu, chorób podstawowych, takich jak cukrzyca, nadciśnienie tętnicze, tarczyca, choroby układu krążenia i nerek, infekcji, a także stosowania leków innych niż rutynowe leki ciążowe (żelazo i suplementy witaminowe). Matka i jej mąż byli kuzynami i nie wspominała o rodzinnym wywiadzie dotyczącym urodzenia noworodka z podobnym zaburzeniem. Prenatalne badania serologiczne były ujemne w kierunku kiły, AIDS, wirusowego zapalenia wątroby typu B i różyczki. Miała grupę krwi B dodatnią.
Według wspomnianego badania ultrasonograficznego wiek ciążowy wynosił 25 tygodni, wskaźnik płynu owodniowego wynosił zero, nerki płodu były niewidoczne w prawidłowym położeniu anatomicznym, pęcherz moczowy był niewidoczny, występowało ciężkie ograniczenie wzrostu płodu i ciężka kardiomegalia (ponad 80%). The diagnosis was Potter syndrome based on ultrasound, after which the mother was taken to the maternity ward.
The mother was hospitalized and, after the normal course of labor, gave birth to a male new-born through vaginal method. The boy had growth limitation (800 g birth weight), the clinical presentation of Potter syndrome, and severe respiratory distress and died immediately after delivery (Figures 1 and 2).
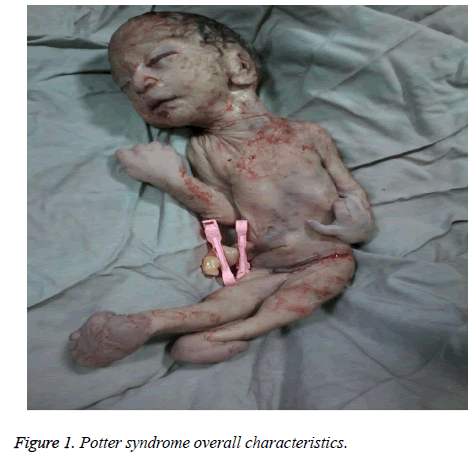
Figure 1: Potter syndrome overall characteristics.
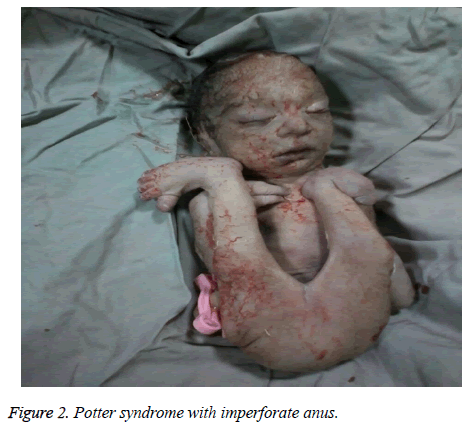
Figure 2: Potter syndrome with imperforate anus.
Discussion
The present study introduced a rare case that has not been reported in the region so far. It was a fatal case of Potter syndrome that was born through normal delivery. Badanie ultrasonograficzne kobiety wykazało płód w 25 tygodniu ciąży z zespołem Pottera i zerowym wskaźnikiem płynu owodniowego. Matka została przyjęta na oddział porodowy, a dziecko urodziło się drogą pochwową. Noworodek z klinicznym obrazem zespołu Pottera miał ciężką niewydolność oddechową i zmarł wkrótce po urodzeniu.
Zespół Pottera, po raz pierwszy opisany przez Edith Potter u noworodków, charakteryzuje się obustronną agenezją nerek lub innymi nieprawidłowościami nerek, takimi jak aplazja, dysplazja, hipoplazja lub wielotorbielowatość nerek. Jest on rozpoznawany częściej u chłopców, a stosunek liczby mężczyzn do liczby kobiet wynosi 2 do 1, co sugeruje obecność pewnych genów na chromosomie Y. Zespół Pottera może być widoczny u noworodków z prawidłowymi nerkami, ale matka miała przewlekły i długotrwały wyciek płynu owodniowego w środkowych tygodniach ciąży .
W tym badaniu, USG kobiet w ciąży ujawniło przypadek zespołu Pottera u płodu płci męskiej w 25 tygodniu ciąży, zerowy wskaźnik płynu owodniowego i brak obu nerek i pęcherza moczowego. Wyniki badań ultrasonograficznych płodów z ciężką chorobą nerek w 23 rodzinach wskazywały na przedłużającą się oligohydramnios, ciężką dysfunkcję nerek i twarz Pottera, a niemowlęta zmarły w ciągu kilku godzin do kilku dni po urodzeniu. Moreira i Reuvers wskazali również na narodziny płodów z zespołem Pottera .
Noworodkowe cechy zespołu Pottera obejmują zmiany twarzy, wady rozwojowe kończyn, ograniczenie wzrostu płodu i hipoplazję płucną, znaną jako oligohydramnios tetrad. Cechy te wynikają z ucisku płodu spowodowanego przedłużającą się oligohydramnios. Twarz potterowska charakteryzuje się hiperteloryzmem, powieką mongolską, fałdem epikantycznym, spłaszczonym mostkiem nosowym i płatkami usznymi, nosem w kształcie dzioba papugi, nisko osadzonymi uszami, wgłębionym podbródkiem, małą fałdą pod wargą, krótką szyją i dodatkowymi łańcuchami na szyi. Prezentacją kliniczną niemowlęcia w tym badaniu była zagłębiona twarz, spłaszczony mostek nosowy i płatki uszne, nos papuzi dziób, nisko osadzone uszy, mały, krótki i zagłębiony podbródek, mała fałda pod wargą, krótka szyja i dodatkowe fałdy wokół szyi, szerokie dłonie, odchylenie dłoni w nadgarstku, obustronna koślawość i poważne ograniczenie wzrostu płodu, co spowodowało masę urodzeniową 800 g. Te objawy kliniczne były zgodne z opisem Shastry i Al-Haggar .
Stały ucisk ścian macicy na ścianę klatki piersiowej płodu oraz ucisk narządów wewnątrzbrzusznych na przeponę stanowią jedną z konsekwencji oligohydramnios w zespole Pottera, które są również głównymi przyczynami hipoplazji i niewydolności płuc w tym zespole. Nasilenie hipoplazji płuc zależy od fazy rozwoju płuc, w której występuje oligohydramnios, a także od intensywności i czasu trwania oligohydramnios. Z powodu ciężkiej niewydolności oddechowej i hipoplazji płuc płody rodzą się z dysplastycznymi nerkami lub umierają wkrótce po urodzeniu. W tym badaniu wskaźnik płynu owodniowego wynosił zero według ultrasonografii, a noworodek miał ciężką niewydolność oddechową przy porodzie i zmarł kilka chwil po urodzeniu.
Zespół Pottera może być związany z wrodzonymi anomaliami serca, anomaliami przewodu pokarmowego (takimi jak atrezja przełyku, agenezja okrężnicy, wady rozwojowe odbytu i dwunastnicy, uchyłek Meckela, torbiele trzustki i śledziony), zaburzeniami układu kostnego, nieprawidłowościami mózgu i różnymi skojarzeniami, takimi jak Vakarl i wsp. W obecnym badaniu ultrasonograficznym odnotowano ciężką kardiomegalię (ponad 80%) i niewidoczny pęcherz moczowy; a badanie fizykalne noworodka ujawniło anomalie kończyn i niedomykalny odbyt.
Główna przyczyna zespołu Pottera pozostaje niejasna w większości przypadków, ale w niektórych przypadkach ma podłoże genetyczne, a wzór dziedziczenia zależy od konkretnej przyczyny genetycznej. Nieprawidłowości genetyczne, takie jak dziedziczenie autosomalne dominujące lub recesywne wielotorbielowatości nerek, dziedzicznej dysplazji nerek, spowodowane mutacjami genów RET i UPK3A oraz nieprawidłowościami chromosomalnymi, mogą powodować nieprawidłowości rozwojowe i prowadzić do zespołu Pottera. Zespół ten występuje sporadycznie, ale może być dziedziczony, gdy powstaje w wyniku triady autosomalnej dominującej. Zespół Pottera jest częstszy u niemowląt, u których w rodzinie występowały nieprawidłowości w budowie nerek. W obecnym badaniu nie znaleziono specyficznej przyczyny tej choroby, a w rodzinie nie występowały anomalie nerek. San i Samal wskazali również na nieznaną etiologię zespołu Pottera i jego przyczyny genetyczne w swoich badaniach .
Chociaż główną przyczyną zespołu Pottera jest nieprawidłowość w rozwoju nerek, wstępna diagnoza jest wykonywana przez USG, które może wykazać utratę lub brak płynu owodniowego i brak pęcherza moczowego, i jest kontynuowana przez dochodzenie w sprawie obecności lub braku nerek. Doradztwo genetyczne jest ważne dla potwierdzenia diagnozy. Rozpoznanie tego zespołu, który charakteryzuje się obustronną agenezją nerek, hipoplazją płuc, twarzą Pottera i wadami rozwojowymi kończyn, może być problematyczne w badaniu ultrasonograficznym z powodu oligohydramnios. W tym badaniu USG wykazało brak wskaźnika płynu owodniowego, niewidoczność pęcherza moczowego oraz obustronną agenezję nerek.
Zespół Pottera jest nie do pogodzenia z życiem i ma śmiertelne rokowanie. Ciąża może być zakończona przed osiągnięciem przez płód stadium życia. Standard opieki nad ciężarną nie ulega zmianie w przypadku podjęcia decyzji o kontynuacji ciąży. Jednym z ograniczeń niniejszego badania było to, że nie przeprowadzono badań genetycznych, ponieważ rodzice nie wyrazili zgody na sekcję zwłok.
W niniejszym badaniu matka w 25 tygodniu ciąży z płodem z zespołem Pottera, rozpoznanym na podstawie badania ultrasonograficznego, została przyjęta na oddział porodowy i urodziła normalnie dziecko, które zmarło kilka chwil po urodzeniu.
Wnioski
Zespół Pottera jest bardzo poważną chorobą i stanem śmiertelnym. Prenatalne badanie ultrasonograficzne pomaga w wykryciu tego zespołu poprzez badanie oligohydramnios i stanu nerek.
Konflikt interesów
Autorzy deklarują, że nie mają konfliktu interesów w związku z publikacją tego opisu przypadku.
Funding
Nie poszukiwano ani nie zapewniono finansowania w związku z tym opisem przypadku.
Zgoda pacjenta
Obtained
Provenance and Peer Review
Ten opis przypadku był recenzowany.
- Garncarz EL. Facial characteristics of infants with bilateral renal agenesis. Am J Obstet Gynecol 1946; 51: 885-888.
- Srihari Babu M, Asha Latha D, Radhika P. Potters syndrome: A case report. JDMS 2015; 14: 14-16.
- Sudhanshu Kumar D, Sidharth Sankar M. Potter facies with polycystic kidney disease in association with other congenital anomalies: two case reports. J Med Dent Sci 2013; 2: 2665-2668.
- Khatami F. Potters syndrome: A study of 15 patients. Arch Iranian Med 2004; 7: 186- 189.
- Sarkar S, Das Gupta S, Barua M, Ghosh R, Mondal K, Chatterjee U, Datta C. Potters sequence: A story of the rare, rarer and the rarest. Indian J Pathol Microbiol 2015; 58: 102-104.
- Manoj MG, Kakkar S. Potters syndrome-A fatal constellation of anomalies. Indian J Med Res 2014; 139: 648-649.
- Himabindu A, Narasinga B. A fatal case of potters syndrome-a case report. JCDR 2011; 5: 1264-1266.
- Moreira BL, Monarim MA, Romano RF, Mattos LA, DIppolito G. Doege-Potter syndrome. Radiol Bras 2015; 48: 195-196.
- Reuvers JR, van Dorp M, Van Schil PE. Solitary fibrous tumor of the pleura with associated Doege-Potter syndrome. Acta Chir Belg 2016; 116: 386-387.
- Shastry SM, Kolte SS, Sanagapati PR. Potters sequence. J Clin Neonatol 2012; 1: 157-159.
- Al-Haggar M, Yahia S, Abdel-Hadi D, Grill F, Al-Kaissi A. Syrenomelia (symelia apus) z zespołem Pottersa w powiązaniu z cukrzycą ciążową: A case report and literature review. Afr Health Sci 2010; 10: 395-399.
- Dhundiraj KM, Madhukar DN, Ambadasrao PG, Wamanrao KS, Prem ZM. Potters syndrome: A report of 5 cases. Indian J Pathol Microbiol 2006; 49: 254-257.
- Dayal J, Maheshwari E, Ghai PS, Bhatotia S. Antenatal ultrasound diagnosis of Potters syndrome. Indian J Radiol Imaging 2003; 13: 81-83.
- San Agustin JT, Klena N, Granath K, Panigrahy A, Stewart E, Devine W, Strittmatter L, Jonassen JA, Liu X. Genetic link between renal birth defects and congenital heart disease. Nat Commun 2016; 22: 111-118.
- Samal SK, Rathod S. Sirenomelia: The mermaid syndrome: report of two cases. J Nat Sci Biol Med 2015; 6: 264-266.
- Gerards FA, Twisk JW, Fetter WP. Predicting pulmonary hypoplasia with 2- or 3-dimensional ultrasonography in complicated pregnancies. Am J Obstet Gynecol 2008; 198: 140-146.
- Kemper MJ, Mueller-Wiefel DE. Prognosis of antenatally diagnosed oligohydramnios of renal origin. Eur J Pediatr 2007; 166: 393-398.