Introducción
El síndrome de regresión caudal (SRC) es un trastorno poco frecuente descrito por primera vez por Geoffroy Saint-Hilaire y Hohl en 1852, y en 1964 Duhmel acuñó el término «síndrome de regresión caudal».1-3 El SRC es un trastorno causado por una anomalía de los segmentos distales de la columna vertebral, y se extiende a una amplia gama de anomalías como la agenesia parcial de la médula espinal, las malformaciones pélvicas asociadas, el ano imperforado, las malformaciones genitales, las anomalías cardíacas, la displasia o aplasia renal bilateral, la hipoplasia pulmonar y la rotación externa extrema con fusión de las articulaciones inferiores que tiene su forma más grave en la sirenomelia (síndrome de la sirena). El SRC también se asocia con hipoplasia femoral, pies deformados y contractura en flexión de las extremidades inferiores. La inteligencia está preservada, en general.1,2,4 Afecta a entre 0,1 y 0,25 de cada 10.000 embarazos, con una proporción hombre-mujer de 2,7:1.3-5 A nivel embrionario, se cree que el SRC es el resultado de defectos en la inducción de elementos caudales en el embrión antes del día 28 de gestación. La lesión se produce en el eje mesodérmico medial posterior provocando la ausencia de desarrollo de la yema mesoblástica caudal.4,6 La etiología exacta es desconocida; sin embargo, se han sugerido como posibles factores la diabetes materna, la predisposición genética y la hipoperfusión vascular.4-7
La diabetes pregestacional es sin duda un teratógeno, y hay buenas evidencias de que la diabetes gestacional puede estar implicada en el desarrollo de la forma más grave de RSC.5
Pinter y reece demostraron que las alteraciones inducidas por la hiperglucemia en el cierre del tubo neural incluyen células desordenadas, disminución de la mitosis y cambios que indican una maduración prematura. El metabolismo oxidativo alterado por la diabetes materna puede provocar un aumento de la producción de radicales libres de oxígeno en el embrión en desarrollo, lo que puede ser teratogénico.8 Entre el 16% y el 22% de los casos de RSC están asociados a la diabetes mellitus materna, lo que aumenta el riesgo de tener un hijo con RSC hasta en un 400.3,4,7 Se han descrito varios casos de familias con RSC, lo que sugiere una posible transmisión genética con diferentes modos de transmisión posibles: Patrones dominantes ligados al X, poligénicos multifactoriales y autosómicos dominantes con penetrancia reducida y expresividad variable.7 La teoría del «robo vascular» fue propuesta inicialmente por Kampmeier en 1927 y reintroducida en 1986 por Stevenson. Adra et al., consideraron la teoría del «robo vascular» de Stevenson como una posible etiología de la patología del SRC.7 Durante la etapa de desarrollo embrionario las estructuras más caudales se separan de los elementos cefálicos como el cerebro, la columna vertebral y la médula espinal, de ahí la falta de alteraciones cognitivas en este síndrome.4
Existen 2 grupos de SRC: el primer grupo es el más afectado con la terminación de la médula espinal por encima de L1. El sacro termina en S1 y, en algunos casos, está ausente. Los pacientes del segundo grupo muestran una disgenesia menos severa con una implantación baja de la médula espinal y atada por un filum terminale engrosado o un lipoma intraespinal.4
Presentación del caso
Presentamos el caso de una recién nacida de una madre de 40 años con antecedentes médicos de miomas uterinos no tratados, antecedentes de ciclos ginecológicos irregulares, 3 embarazos con 2 partos y colecistectomía en 2008. Menciona un embarazo no planificado pero deseado, con un control prenatal de alto riesgo. Acudió a más de 15 consultas prenatales. En la semana 18 de gestación se encontraron datos compatibles con malformaciones del tubo neural con agenesia sacra, lo que dio lugar a un diagnóstico de RSC. Durante el segundo trimestre de gestación se detectó hiperglucemia, por lo que se le puso en tratamiento con insulina. A las 40 semanas de gestación se realizó cesárea obteniendo un producto sano, con una puntuación de Apgar de 8/9 con una edad gestacional de 39,6 semanas. En la exploración física observamos un peso de 3,44 kg, una longitud de 49 cm, un perímetro cefálico de 35 cm y un perímetro abdominal de 32 cm. Durante la inspección pudimos observar una evidente fetopatía diabética, con abundante pelo, y baja implantación (Fig. 1). Fontanela anterior normal (2 x 2 cm), cara redonda, pómulos prominentes, fisuras palpebrales horizontales, puente nasal corto, punta redonda, labios finos, paladar lleno, displasia de los pabellones auriculares con hipertricosis del hélix, cuello corto con abultamiento dorsal, tórax normal, sin soplos durante la auscultación, el abdomen era blando sin masas palpables ni visceromegalias, columna vertebral lineal con presencia de un hoyuelo en la piel en la región lumbosacra (Fig. 2), no se palpaba ningún hueso del sacro, posición normal del ano, permeable, con un tono ausente. Había un evidente acortamiento de las extremidades inferiores y pie varo bilateral (Fig. 3). Se realizaron radiografías para ver las malformaciones óseas, pudiendo observar una agenesia completa del sacro con una fusión lumbo-ilíaca. Una resonancia magnética (RM) cerebral y de la columna vertebral (Fig. 4), informó de una terminación abrupta de la cuerda y de la zona lumbar a nivel de L3. Después de este nivel sólo se identifican masas amorfas con intensidad de señal grasa, relacionadas con el tejido subcutáneo. Médula espinal corta, cono medular plano a nivel de T10, ambos huesos ilíacos eran hipoplásicos y estaban fusionados a nivel medio. No podemos ver el sacro. Se pudieron ver ambos riñones malrotados e hipoplásicos, una vejiga urinaria prominente compatible con una vejiga neurogénica. La imagen del cerebro fue reportada como normal. Se solicitó una ecografía de abdomen superior para descartar otras anomalías viscerales encontrando una discrepancia en el tamaño del riñón izquierdo, además de no encontrar su configuración normal, lo que sugería una discreta hipoplasia así como una posible malrotación. Durante su estancia se realizó consulta multidisciplinar, y se colocaron férulas de yeso en ambos miembros inferiores. El paciente permanece en seguimiento con Urología, Traumatología, Neurología y Pediatría General, esto con el fin de mantener una estrecha vigilancia de todos los factores de riesgo y posibles complicaciones.
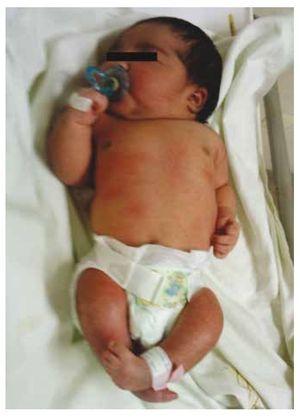
Figura 1 Recién nacido con fetopatía diabética.
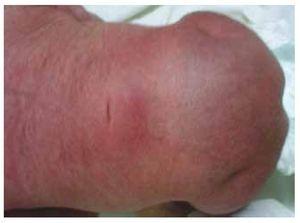
Figura 2 Un hoyuelo en la piel en la región lumbosacra.
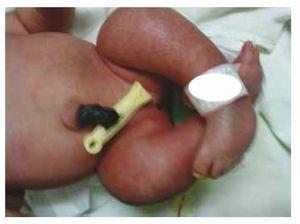
Figure 3 Equinovarus feet.
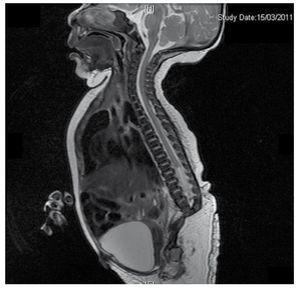
Figure 4 The lumbar spine magnetic resonance imaging (MRI) shows an abrupt chord and lumbar termination at the L3 level. Short spinal cord, flat conus medullaris at T10 level, hypoplastic iliac bones, fused at mid-level. No sacrum is seen.
Discussion
The CRS is a rare congenital malformation. Even though the specific etiologic factor is unknown, it is related to maternal diabetes, genetic predisposition and vascular hypoperfusion.3 Just as in the case presented, this alteration is characterized by agenesis of the sacrum involving iliac and lumbar vertebrae with their corresponding spinal segments and variable abnormalities in the lower limbs as well as in other organs. Fetal diagnosis tools allow early syndrome detection. La ecografía prenatal es el instrumento paraclínico más utilizado; un elemento clave de la ecografía prenatal es la evaluación detallada de la columna vertebral y las extremidades inferiores; también permite el diagnóstico del SRC mediante la demostración de una terminación abrupta de la columna lumbar y de extremidades inferiores hipoplásicas. Al realizar el diagnóstico prenatal, debemos centrarnos en discernir el grado de digénesis así como las anomalías congénitas asociadas con el fin de establecer un pronóstico y un plan oportuno de intervenciones terapéuticas postnatales.3,5 La clasificación de Renshav, creada en 1978, clasifica el síndrome en 4 grados según la gravedad de la agenesia del sacro y la afectación de las vértebras ilíacas y lumbares.9 Según esta clasificación, nuestra paciente pertenece al grado iV (agenesia completa del sacro con fusión de huesos ilíacos). Este grupo se asocia a un pronóstico aún peor con una mayor repercusión neurológica y secuelas multisistémicas, principalmente a nivel renal. Este caso es un claro ejemplo del amplio abanico de alteraciones que pueden afectar al feto en crecimiento como consecuencia de una diabetes materna no controlada durante el embarazo. Debido a la alta correlación entre este defecto y la madre diabética, y su desarrollo durante las primeras etapas del embarazo, es imprescindible una estrategia preventiva que incluya un estricto control glucémico antes del periodo de organogénesis embrionaria, o incluso antes en pacientes de alto riesgo. También son importantes la orientación adecuada y los exámenes genéticos pregestacionales. El tratamiento es un reto tanto para el médico como para los padres, y exige un enfoque multidisciplinar que implique a un pediatra, un cirujano pediátrico, un cirujano ortopédico, un fisioterapeuta y un urólogo en función de la gravedad. Given the fact that the primary pathology is irreversible, treatment is just supportive, with the sole purpose of accomplishing a life as normal as possible.
Conflicts of interest
The authors have no conflicts of interest to declare.
Funding
No financial support was provided.
Received: September 2013;
Accepted: January 2014