Case Report – Biomedical Research (2019) Volume 30, Issue 2
Report of a deadly case of potter syndrome: Een case report.
Mehrbano Amirshahi, Mahin Badakhsh* en Zohreh Sadat Hashemi
Department of Midwififery, School of Nursing and Midwifery, Zabol University of Medical sciences, Zabol, Iran
*Corresponderende Auteur: Mahin Badakhsh
Department of Midwifery
School of Nursing and Midwifery
Zabol University of Medical sciences
Iran
Accepted date: 21 februari 2019
Abstract
Achtergronden: Het syndroom van Potter is een zeldzame aangeboren aandoening die verwijst naar een reeks klinische manifestaties die gepaard gaan met oligohydramnios veroorzaakt door foetaal nierfalen. Het kenmerk van dit syndroom is het bijzondere klinische beeld dat naast oligohydramnios kenmerkend is voor pulmonale hypoplasie, bilaterale renale agenese; misvormingen van ledematen en een specifiek gezicht. Het embryo sterft voor of onmiddellijk na de geboorte ten gevolge van ademhalingsmoeilijkheden. Het doel van deze studie is verslag uit te brengen van een foetus met het syndroom van Potter die vaginaal ter wereld kwam. Casus verslag: Uit echografisch onderzoek van de zwangere vrouw bleek dat haar mannelijke foetus met een zwangerschapsduur van 25 weken met het syndroom van Potter was geboren en dat de vruchtwaterindex nul is. De moeder werd opgenomen in het ziekenhuis in unit labour en beviel in een natuurlijk verloop. De baby had een klinisch beeld van het syndroom van Potter en ernstige ademnood en overleed kort na de geboorte. Conclusie: Het syndroom van Potter is een zeer ernstige aandoening die meestal dodelijk is. Prenatale echografie door onderzoek van oligohydramnios en nieren helpt bij het stellen van de diagnose.
Keywords
Potter’s syndroom, Oligohydramnios, Pulmonale hypoplasie, Foetus
Introductie
Potter syndroom is een zeldzame aangeboren afwijking die voornamelijk mannelijke foetussen treft en wordt gekenmerkt door pulmonale hypoplasie veroorzaakt door nierfalen. Het werd voor het eerst gemeld door Edith Potter in 1946. Na de 16e week van de zwangerschap hangt de hoeveelheid vruchtwater voornamelijk af van de urineproductie van de foetus. Tijdens het intra-uteriene leven slikt de foetus voortdurend het vruchtwater in dat door de nieren weer naar de vruchtzak wordt teruggevoerd. Oligohydramnio treedt op wanneer het volume van het vruchtwater minder is dan normaal voor die periode van de zwangerschap. Deze volumevermindering kan te wijten zijn aan een verminderde urineproductie ten gevolge van oorzaken zoals bilaterale nierabesitas, obstructie van de urinewegen en het langdurig ruptureren van de vruchtzak. De urine van de foetus, die essentieel is voor de ontwikkeling van de longen, draagt bij tot de ontwikkeling van de luchtwegen en de alveoli, creëert hydraulische druk en levert proline, een essentieel aminozuur voor de ontwikkeling van de longen. Als de longblaasjes en bijgevolg de longen bij de geboorte niet voldoende ontwikkeld zijn, zal de pasgeborene niet goed kunnen ademen en lijden aan ademnood ten gevolge van pulmonale hypoplasie. Daarom is longhypoplasie, secundair aan nierfalen, de belangrijkste doodsoorzaak bij pasgeborenen met het Potter-syndroom.
De productie van urine door het embryo beïnvloedt niet alleen het volume van het vruchtwater, maar beschermt het embryo ook tegen de druk van de baarmoederwand van de moeder, als een kussen. Oligohydramnios resulteert in een speciale vorm van de foetus, “Potter facies” genoemd, die wordt gekenmerkt door kenmerken zoals een afgeplatte neusbrug, hypoplastische kin, epicanthal plooien, cataract, en laag aangezette oren.
De belangrijkste oorzaak van het Potter syndroom is onbekend; dit syndroom heeft in sommige gevallen een genetische achtergrond, en komt vaker voor bij neonaten met een familiegeschiedenis van nierafwijkingen . Het syndroom, met een incidentie van 1 op de 2.000 tot 5.000 foetussen, wordt geassocieerd met een recidief risico van 3-6%, en wordt gevonden in 0,2-0,4% van de autopsies bij dode pasgeborenen of degenen die direct na de geboorte overlijden . Er wordt echter aangenomen dat de ziekte een hogere prevalentie heeft omdat de getroffen foetussen dood geboren worden of kort na de geboorte sterven. Er is geen methode bekend om deze dodelijke ziekte te voorkomen. Daarom wordt bij moeders met een positieve zwangerschapsanamnese voor dit syndroom een screeningsechografie bij 16-18 weken zwangerschap aanbevolen om oligohydramnios en de nieren van de foetus te evalueren. Hoewel dit syndroom dodelijke gevolgen heeft en niet verenigbaar is met het leven en eindigt met de dood van de baby, vereisen de getroffen foetussen reanimatie en behandeling van de urinewegobstructie bij de bevalling. De huidige studie had tot doel verslag te doen van een foetus met het syndroom van Potter die via een normale bevalling werd geboren.
Casusverslag
Het geval was een 25 weken oude mannelijke foetus met het syndroom van Potter, geboren via een normale bevalling. De moeder werd op 10 mei 2017 opgenomen op de kraamafdeling van het Amir al-Momenin Ziekenhuis in Zabol, Iran, met een echografie, waaruit de afwezigheid van vruchtwater en de diagnose Potter-syndroom voor de foetus bleek. De moeder was 24 jaar, Iraanse, huisvrouw, gravid 3, en para 1, en had een abortusgeschiedenis bij maand 1. Ze herinnerde zich de eerste dag van haar laatste menstruatie niet, en haar zwangerschapsleeftijd was 25 weken volgens de enige echografie die op dezelfde dag werd uitgevoerd. Ze had een normale bevallingsgeschiedenis (haar vorige kind was gezond en had een normale bevalling gehad), en had prenatale zorg ontvangen tijdens de zwangerschap. Haar zwangerschapstesten waren normaal. Er was geen voorgeschiedenis van roken, drugs of alcohol, onderliggende ziekten zoals diabetes, hypertensie, schildklier, cardiopulmonale en renale aandoeningen, infectie, en medicijngebruik anders dan routine zwangerschapsmedicijnen (ijzer- en vitaminesupplementen). De moeder en haar echtgenoot waren neef en nicht en zij vermeldde geen familiegeschiedenis van een neonatale geboorte met een gelijkaardige aandoening. Prenatale serologische tests waren negatief voor syfilis, AIDS, hepatitis B, en rodehond. Ze had bloedgroep B positief.
Volgens de genoemde echografie was de zwangerschapsduur 25 weken, de vruchtwaterindex nul, waren de nieren van de foetus onzichtbaar in de normale anatomische positie, was de blaas onzichtbaar, en was er sprake van ernstige foetale groeibeperking en ernstige cardiomegalie (meer dan 80%). The diagnosis was Potter syndrome based on ultrasound, after which the mother was taken to the maternity ward.
The mother was hospitalized and, after the normal course of labor, gave birth to a male new-born through vaginal method. The boy had growth limitation (800 g birth weight), the clinical presentation of Potter syndrome, and severe respiratory distress and died immediately after delivery (Figures 1 and 2).
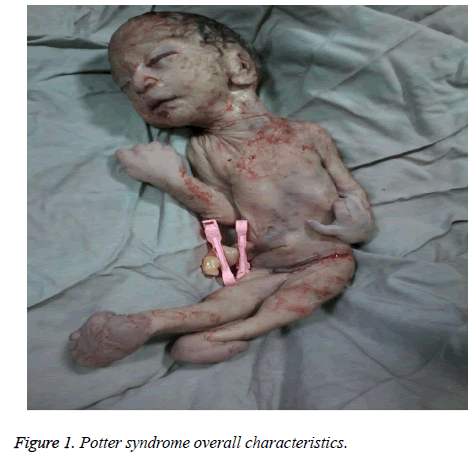
Figure 1: Potter syndrome overall characteristics.
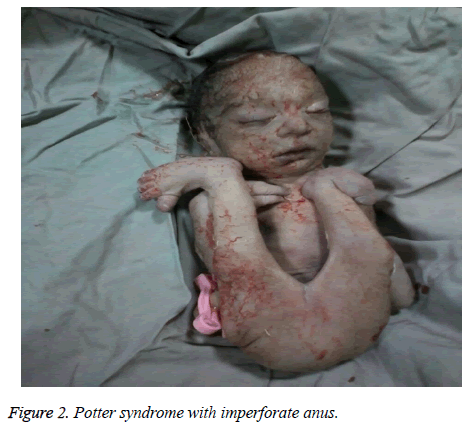
Figure 2: Potter syndrome with imperforate anus.
Discussion
The present study introduced a rare case that has not been reported in the region so far. It was a fatal case of Potter syndrome that was born through normal delivery. De echografie van de vrouw toonde een foetus bij 25 weken zwangerschap met het syndroom van Potter en een vruchtwaterindex van nul. De moeder werd opgenomen op de verlosafdeling en de baby werd vaginaal geboren. De pasgeborene met de klinische presentatie van het syndroom van Potter had ernstige ademnood en overleed kort na de geboorte.
Potter syndroom, voor het eerst beschreven door Edith Potter bij pasgeborenen, wordt gekenmerkt door bilaterale renale agenese of andere nierafwijkingen zoals aplasie, dysplasie, hypoplasie, of polycysteuze nierziekte. Het wordt meer herkend bij jongens met een man-vrouw verhouding van 2 op 1, wat wijst op de aanwezigheid van bepaalde genen op chromosoom Y . Het syndroom van Potter kan worden gezien bij pasgeborenen met normale nieren, maar de moeder had een chronische en langdurige vruchtwaterlekkage tijdens de middelste weken van de zwangerschap .
In deze studie onthulde de echografie van de zwangere vrouwen een geval van het syndroom van Potter bij een mannelijke foetus in de 25e week van de zwangerschap, een vruchtwaterindex van nul, en het ontbreken van zowel nieren als blaas. Echografische bevindingen van foetussen met ernstige nieraandoeningen in 23 families wezen op langdurige oligohydramnios, ernstige nierdisfunctie, en Potter-facies, en de baby’s stierven binnen enkele uren tot dagen na de geboorte. Moreira en Reuvers wezen ook op de geboorte van foetussen met het syndroom van Potter.
De neonatale kenmerken van het syndroom van Potter omvatten gezichtsveranderingen, misvormingen van ledematen, foetale groeibeperking, en pulmonale hypoplasie, bekend als oligohydramnios tetrad. Deze kenmerken ontstaan door foetale compressie als gevolg van langdurige oligohydramnios. Het Potter-gezicht wordt gekenmerkt door hypertelorisme, een mongools ooglid, een epicanthusplooi, een afgeplatte neusbrug en oorlel, een papegaaienbekneus, laag aangezette oren, een teruggetrokken kin, een kleine plooi onder de lip, een korte hals en extra kettingen rond de hals. De klinische presentatie van het kind in deze studie was een teruggetrokken gezicht, een afgeplatte neusbrug en oorlel, een papegaaienbekneus, laag aangezette oren, een kleine, korte en teruggetrokken kin, een kleine plooi onder de lip, een korte hals, en extra plooien rond de hals, brede handen, handafwijking bij de pols, bilaterale valgus, en ernstige foetale groeibeperking, wat resulteerde in een geboortegewicht van 800 g. Deze klinische manifestaties waren consistent met Shastry en Al-Haggar .
De constante druk van de baarmoederwanden op de borstwand van de foetus en de druk van intra-abdominale organen op het diafragma vormen een van de gevolgen van oligohydramnios bij het Potter syndroom, die tevens de belangrijkste oorzaken zijn van longhypoplasie en -falen bij dit syndroom. De ernst van de pulmonale hypoplasie hangt af van de ontwikkelingsfase van de longen waarin oligohydramnio optreedt, alsook van de intensiteit en de duur van de oligohydramnio. Als gevolg van ernstige ademnood en longhypoplasie worden foetussen geboren met dysplastische nieren of sterven ze kort na de geboorte. In deze studie was de vruchtwaterindex nul volgens de echografie en de pasgeborene had ernstige ademnood bij de geboorte en overleed enkele ogenblikken na de geboorte.
Potter syndroom kan geassocieerd worden met aangeboren hartafwijkingen, gastro-intestinale afwijkingen (zoals slokdarm atresie, colon agenese, en anale en duodenale misvormingen, Meckel’s divertikel, en pancreas en milt cysten), skeletafwijkingen, hersenafwijkingen, en verschillende associaties, zoals Vakarl et al. . In de huidige studie werd bij de echografie een ernstige cardiomegalie (meer dan 80%) en onzichtbare blaas vastgesteld; en bij het lichamelijk onderzoek van de pasgeborene werden anomalieën van de ledematen en een imperforate anus vastgesteld.
De hoofdoorzaak van het Potter-syndroom blijft in de meeste gevallen onduidelijk, maar het heeft in sommige gevallen een genetische oorzaak, en het overervingspatroon is afhankelijk van een bepaalde genetische oorzaak. Genetische afwijkingen zoals autosomaal dominante of recessieve overerving van polycysteuze nierziekte, erfelijke nierdysplasie, veroorzaakt door RET en UPK3A genmutaties en chromosomale afwijkingen, kunnen resulteren in ontwikkelingsafwijkingen en leiden tot het syndroom van Potter. Dit syndroom komt sporadisch voor, maar kan erfelijk zijn wanneer het voortkomt uit de autosomaal dominante trias. Het syndroom van Potter komt vaker voor bij zuigelingen met een familiegeschiedenis van nierafwijkingen. In de huidige studie werd geen specifieke oorzaak gevonden voor deze ziekte, en er was geen familiegeschiedenis van nierafwijkingen. San en Samal wezen ook op de onbekende etiologie van het syndroom van Potter en de genetische redenen in hun studies .
Hoewel de belangrijkste oorzaak van het syndroom van Potter de afwijking in de ontwikkeling van de nieren is, wordt de initiële diagnose uitgevoerd door echografie die het verlies of de afwezigheid van vruchtwater en de afwezigheid van blaas kan aantonen, en wordt vervolgd door onderzoek naar de aanwezigheid of afwezigheid van nieren. Genetisch advies is belangrijk om de diagnose te bevestigen. De identificatie van dit syndroom, dat wordt gekenmerkt door bilaterale renale agenese, pulmonale hypoplasie, Potter-facies en misvorming van ledematen, kan problematisch zijn door echografie als gevolg van oligohydramnios . In deze studie toonde de echografie de afwezigheid van vruchtwater index, onzichtbaarheid van de blaas, en bilaterale renale agenese.
Potter syndroom is onverenigbaar met het leven en heeft een dodelijke prognose. De zwangerschap kan worden afgebroken voordat de foetus het levensstadium bereikt. De standaard zwangerschapszorg verandert niet wanneer besloten wordt de zwangerschap voort te zetten . Een van de beperkingen van de huidige studie was dat genetisch onderzoek niet werd uitgevoerd, omdat de ouders geen autopsie accepteerden.
In de huidige studie werd een moeder in haar 25e week van de zwangerschap met een foetus met het Potter-syndroom, gediagnosticeerd door middel van echografie, opgenomen in de verlosafdeling en gaf een normale geboorte aan een baby die enkele ogenblikken na de geboorte overleed.
Conclusie
Potter-syndroom is een zeer ernstige ziekte en een dodelijke aandoening. Prenatale echografie helpt dit syndroom op te sporen door oligohydramnios en nieraandoeningen te onderzoeken.
Conflict of Interest
De auteurs verklaren dat zij geen belangenconflict hebben met betrekking tot de publicatie van dit case report.
Funding
No funding was sought or secured in relation to this case report.
Patient Consent
Obtained
Provenance and Peer Review
Dit case report was peer reviewed.
- Potter EL. Facial characteristics of infants with bilateral renal agenesis. Am J Obstet Gynecol 1946; 51: 885-888.
- Srihari Babu M, Asha Latha D, Radhika P. Potters syndroom: A case report. JDMS 2015; 14: 14-16.
- Sudhanshu Kumar D, Sidharth Sankar M. Potter facies met polycysteuze nierziekte in associatie met andere congenitale afwijkingen: twee case reports. J Med Dent Sci 2013; 2: 2665-2668.
- Khatami F. Potters syndroom: Een studie van 15 patiënten. Arch Iranian Med 2004; 7: 186- 189.
- Sarkar S, Das Gupta S, Barua M, Ghosh R, Mondal K, Chatterjee U, Datta C. Potters sequence: Een verhaal van het zeldzame, zeldzamere en het zeldzaamste. Indian J Pathol Microbiol 2015; 58: 102-104.
- Manoj MG, Kakkar S. Potters syndroom-een fatale constellatie van anomalieën. Indian J Med Res 2014; 139: 648-649.
- Himabindu A, Narasinga B. Een fataal geval van potters syndroom-een case report. JCDR 2011; 5: 1264-1266.
- Moreira BL, Monarim MA, Romano RF, Mattos LA, DIppolito G. Doege-Potter syndroom. Radiol Bras 2015; 48: 195-196.
- Reuvers JR, van Dorp M, Van Schil PE. Solitaire fibreuze tumor van het borstvlies met geassocieerd Doege-Potter syndroom. Acta Chir Belg 2016; 116: 386-387.
- Shastry SM, Kolte SS, Sanagapati PR. Potters sequentie. J Clin Neonatol 2012; 1: 157-159.
- Al-Haggar M, Yahia S, Abdel-Hadi D, Grill F, Al-Kaissi A. Sirenomelie (symelia apus) met Potters syndroom in verband met zwangerschapsdiabetes mellitus: A case report and literature review. Afr Health Sci 2010; 10: 395-399.
- Dhundiraj KM, Madhukar DN, Ambadasrao PG, Wamanrao KS, Prem ZM. Potters syndroom: Een verslag van 5 gevallen. Indian J Pathol Microbiol 2006; 49: 254-257.
- Dayal J, Maheshwari E, Ghai PS, Bhatotia S. Antenatale echografische diagnose van het syndroom van Potters. Indian J Radiol Imaging 2003; 13: 81-83.
- San Agustin JT, Klena N, Granath K, Panigrahy A, Stewart E, Devine W, Strittmatter L, Jonassen JA, Liu X. Genetisch verband tussen niergeboorteafwijkingen en aangeboren hartaandoeningen. Nat Commun 2016; 22: 111-118.
- Samal SK, Rathod S. Sirenomelia: The mermaid syndrome: report of two cases. J Nat Sci Biol Med 2015; 6: 264-266.
- Gerards FA, Twisk JW, Fetter WP. Predicting pulmonary hypoplasia with 2- or 3-dimensional ultrasonography in complicated pregnancies. Am J Obstet Gynecol 2008; 198: 140-146.
- Kemper MJ, Mueller-Wiefel DE. Prognosis of antenatally diagnosed oligohydramnios of renal origin. Eur J Pediatr 2007; 166: 393-398.